Protein Crystallization, Hanging and Sitting Drops
Preparing the protein sample
Protein crystallization starts from protein production (cloning, expression & purification) and subsequent thorough characterization of the purified protein in solution. For crystallization, the protein should be of high purity (better than 99%), stable and monodisperse. Monodisperse means that the solution should not contain aggregates or denatured material and preferably should not contain a variety of oligomeric forms of the protein. We also want it to be stable within the pH range and other buffer conditions we plan to use in subsequent work. On our company website, you can find a more detailed description of the process of protein sample preparation for crystallization.
Thermodynamics of crystal formation
For a protein to crystallize, we must bring the solution into a supersaturated state. It is like getting crystals of regular table salt - we can heat the solution in a cup and try to dissolve as much salt as possible to get a saturated solution. Then we let the cup with the solution stand on the table until its temperature goes down to room temperature. The solution is now supersaturated since salt solubility at room temperature is lower than solubility at an elevated temperature. While standing on the table, tiny salt crystals may appear at the cup's bottom. If the solution is very "clean," which means no small dust particles or anything similar in the cup (impurities may promote nucleation and crystal growth), the solution may stay clear on the table for a while. However, since the supersaturated solution is thermodynamically unstable, even some external factors, like a slight vibration of the table, may start the crystallization process, which is a transition from the supersaturated state to a "normal" saturated state.
The simple technique for getting salt crystals (by dissolving salt in hot water) cannot be used for protein crystallization. However, several other methods and tools have been developed to handle protein solutions and bring them into a supersaturated state. The easiest is the so-called hanging and sitting drop methods which are shown schematically in the image below:
Protein crystallization starts from protein production (cloning, expression & purification) and subsequent thorough characterization of the purified protein in solution. For crystallization, the protein should be of high purity (better than 99%), stable and monodisperse. Monodisperse means that the solution should not contain aggregates or denatured material and preferably should not contain a variety of oligomeric forms of the protein. We also want it to be stable within the pH range and other buffer conditions we plan to use in subsequent work. On our company website, you can find a more detailed description of the process of protein sample preparation for crystallization.
Thermodynamics of crystal formation
For a protein to crystallize, we must bring the solution into a supersaturated state. It is like getting crystals of regular table salt - we can heat the solution in a cup and try to dissolve as much salt as possible to get a saturated solution. Then we let the cup with the solution stand on the table until its temperature goes down to room temperature. The solution is now supersaturated since salt solubility at room temperature is lower than solubility at an elevated temperature. While standing on the table, tiny salt crystals may appear at the cup's bottom. If the solution is very "clean," which means no small dust particles or anything similar in the cup (impurities may promote nucleation and crystal growth), the solution may stay clear on the table for a while. However, since the supersaturated solution is thermodynamically unstable, even some external factors, like a slight vibration of the table, may start the crystallization process, which is a transition from the supersaturated state to a "normal" saturated state.
The simple technique for getting salt crystals (by dissolving salt in hot water) cannot be used for protein crystallization. However, several other methods and tools have been developed to handle protein solutions and bring them into a supersaturated state. The easiest is the so-called hanging and sitting drop methods which are shown schematically in the image below:
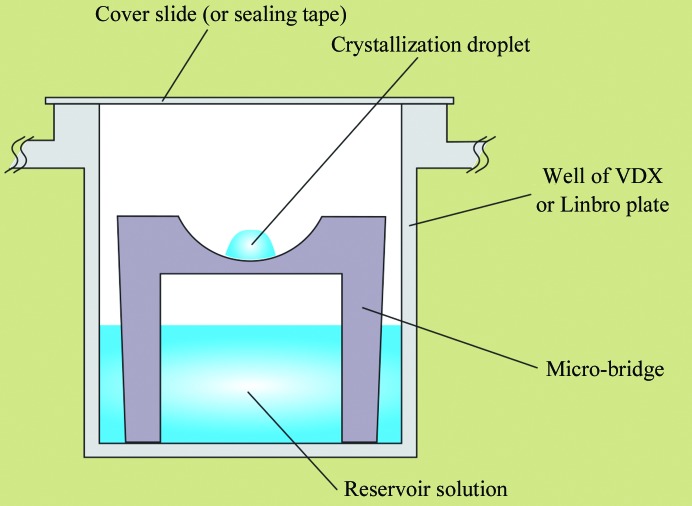
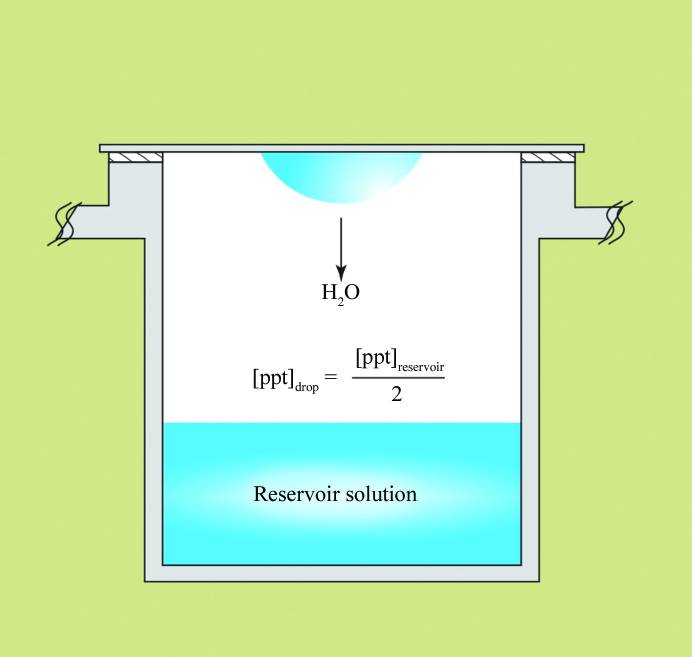
Sitting and hanging drop crystallization. Image from McPherson & Gavira, 2014. There are, of course, many other methods used for creating a concentration gradient for promoting supersaturation.
The vapor diffusion methods, based on hanging or sitting drops (images above), are the most frequently used in protein crystallization. Usually, the precipitant concentration in the reservoir is higher than in the drop. Due to the concentration gradient, the water from the drop will evaporate toward the reservoir, thus lowering the volume of the drop and increasing the protein concentration. At some moment, the concentration of the protein will reach supersaturation. If all other parameters, like pH, ionic strength, type of buffer, additives, etc., are appropriate, the protein may start to form crystals (crystallize).





Images showing growing crystals and a 24-well crystallization plate.
The crystal gallery at Hampton research includes a lot of beautiful crystal images!
The crystal gallery at Hampton research includes a lot of beautiful crystal images!
Screening for crystallization conditions
Suppose we have a new protein to crystallize and don't know the exact conditions for crystallization (like the type of buffer, pH, salt concentration, etc.). In that case, we need to screen for crystallization conditions. At the start, we need to test many combinations with different duffers, different salts and salt concentrations, the effect of ligands, and different precipitants. The precipitant is the main component responsible for the creation of the "concentration gradient" required in vapor diffusion and other types of crystallization methods. Many different kinds of precipitants and ionic liquids are used in protein crystallography; probably the most common are ammonium sulfate, polyethylene glycols of various lengths, and propanol. Below is a table listing some common factors that affect protein solubility. I have also downloaded an example of a screen offered by Hampton Research to show the large number of salts and precipitants used in this screen alone. And there are a lot of other screens! This should give an idea of how protein crystallography works - many ready-made screens are tested before the right crystallization conditions are found! We can purchase the crystallization screens and set up hanging or sitting drops with hundreds of conditions.
Specialized liquid handling robotics is used to set up nano-liter sitting-drop crystallization screens. An example is the Mosquito liquid handling robot that is used with 96-well plates. A liquid handling robot may need as little as 15 microL of protein sample to screen 96 different crystallization conditions. The process is speedy and takes a couple of minutes. However, the initial conditions found from these screens often need to be further optimized before crystals useful for X-ray data collection are produced.
The protein concentration to be used in screening depends on the solubility of the protein. As a rule of thumb, the higher the solubility of the proteins, the higher the concentration should be. Remember that we need to reach a supersaturation level! The range between 5 to 10 mg/ml is expected for most proteins. It is also possible to assess the required initial concentration using a so-called pre-crystallization test (PCT), for example, from Hampton Research.
X-ray data collection
Grown crystals must be removed from the drops to mount them into an X-ray beam. To protect the crystals from radiation damage caused by high-intensity X-rays at synchrotrons, crystals are frozen at a liquid nitrogen temperature, and X-ray data are collected at these temperatures (cryo-temperatures). Sometimes data from crystals at room temperature are collected directly using the crystallization plates placed into an X-ray beam at a synchrotron. However, many crystals are required for this to work since each crystal may survive only one single shot of the high-intensity synchrotron beam. The next page gives some details of the X-ray crystallography experiment.
Suppose we have a new protein to crystallize and don't know the exact conditions for crystallization (like the type of buffer, pH, salt concentration, etc.). In that case, we need to screen for crystallization conditions. At the start, we need to test many combinations with different duffers, different salts and salt concentrations, the effect of ligands, and different precipitants. The precipitant is the main component responsible for the creation of the "concentration gradient" required in vapor diffusion and other types of crystallization methods. Many different kinds of precipitants and ionic liquids are used in protein crystallography; probably the most common are ammonium sulfate, polyethylene glycols of various lengths, and propanol. Below is a table listing some common factors that affect protein solubility. I have also downloaded an example of a screen offered by Hampton Research to show the large number of salts and precipitants used in this screen alone. And there are a lot of other screens! This should give an idea of how protein crystallography works - many ready-made screens are tested before the right crystallization conditions are found! We can purchase the crystallization screens and set up hanging or sitting drops with hundreds of conditions.
Specialized liquid handling robotics is used to set up nano-liter sitting-drop crystallization screens. An example is the Mosquito liquid handling robot that is used with 96-well plates. A liquid handling robot may need as little as 15 microL of protein sample to screen 96 different crystallization conditions. The process is speedy and takes a couple of minutes. However, the initial conditions found from these screens often need to be further optimized before crystals useful for X-ray data collection are produced.
The protein concentration to be used in screening depends on the solubility of the protein. As a rule of thumb, the higher the solubility of the proteins, the higher the concentration should be. Remember that we need to reach a supersaturation level! The range between 5 to 10 mg/ml is expected for most proteins. It is also possible to assess the required initial concentration using a so-called pre-crystallization test (PCT), for example, from Hampton Research.
X-ray data collection
Grown crystals must be removed from the drops to mount them into an X-ray beam. To protect the crystals from radiation damage caused by high-intensity X-rays at synchrotrons, crystals are frozen at a liquid nitrogen temperature, and X-ray data are collected at these temperatures (cryo-temperatures). Sometimes data from crystals at room temperature are collected directly using the crystallization plates placed into an X-ray beam at a synchrotron. However, many crystals are required for this to work since each crystal may survive only one single shot of the high-intensity synchrotron beam. The next page gives some details of the X-ray crystallography experiment.
Common factors that affect protein solubility
1) pH
2) Ionic strength
3) Concentration of precipitant
4) Concentration of macromolecule
5) Temperature
6) Additives, effectors, and ligands
8) Organism source of macromolecules
9) Presence of substrates, coenzymes, inhibitors
10) Reducing or oxidizing environment
11) Metal ions
12) Rate of equilibration
Even gravity has been considered as one of the possible crystallization parameters!
The list may be extended, and many more factors affecting protein solubility may be added. Commercially available crystallization screens include hundreds of solutions with combinations of pH, ionic strength, salts, precipitants, so-called additives (small molecules that may promote crystallization), metal ions, etc.