Sequence Alignment Tools: Databases, Multiple, Global & Local Alignment
This chapter focuses on understanding multiple protein sequence alignment, sequence databases, and the tools for multiple alignment and analysis.
Understanding protein sequence alignment and thes essential for any sequence analysis, although studying it can be challenging and requires prior knowledge and experience. We can sometimes compare such a study to trying to read a text in a foreign language without understanding it: we may recognize the letters and even some words, but we cannot grasp the meaning of the text. Since our focus here is on structural biology and the relationships between amino acid sequences, structures, and functions, I will only present an overview of the basic concepts and techniques used in sequence alignment and analysis without delving into the theory and algorithms behind the process.
Before running sequence alignment and analysis, we need to understand the essential characteristics of the 20 most common amino acids that build up the proteins we find in nature. The amino acids can be classified into three main groups: hydrophobic, polar, and charged. Different amino acids are responsible for different functions in proteins. Some are often found at functional sites; hydrophobic amino acids usually build up the core of protein molecules, while charged residues are distributed on the protein surface in contact with a solvent. A sequence alignment will quickly reveal the distribution of these residues along the protein amino acid sequence and will also show their conservation pattern in a protein family.
Many positions in a protein sequence change during evolution due to mutations in the genetic code. However, many of these substitutions are so-called conserved substitutions. This means that other hydrophobic amino acids usually replace hydrophobic amino acids, which also applies to charged and polar amino acids. In other words, we observe the conservation of the chemical nature of an amino acid. This raises the question of how we account for these substitutions when running and scoring a sequence alignment. Mutation probability matrices (substitution matrices) were introduced to answer this question. These matrices consider the frequency of amino acid substitution replacements in calculating an alignment score.
The first step in sequence alignment and analysis is placing the protein we are interested in within a specific frame, which is the protein family to which it belongs. Within a family, proteins perform a similar function, have conserved sequence features, conserved three-dimensional structures, etc. An alignment will help us reveal all these characteristic features, find the conserved and variable regions in the sequence, show functionally essential residues, extract information on the secondary and tertiary structure, etc. This chapter will discuss the techniques used to perform sequence alignment and analysis.
For an alignment, we first need to find related sequences. To do this, we need to run a database search. The software will compare our sequence (query sequence) to all other sequences in the database by using a local alignment algorithm BLAST and, based on specific criteria, will output several amino acid sequences found to be related to our protein. Local alignment compares small segments of the query sequence to other sequences in the database to find matching amino acid segments. If we are happy with the search results and have a list of proteins to start the analysis with, we can run a so-called global alignment. In this case, we can run a pair-wise alignment of only two sequences or a multiple sequence alignment (MSA) using a group of sequences. MSA constitutes the basis of any sequence analysis and provides much more information than the alignment of a pair of sequences. It is extensively used in secondary and tertiary structure prediction and modeling to identify protein families and domains. There are many sequence alignment and analysis tools on the web. You will find an example in the sequence alignment tutorial that will guide you through the jungle.
The image below shows an example of a BLAST search. I usually use the Expasy and EBI servers. Local alignment compares small segments of the query sequence to other sequences in the database to find matching amino acid segments. This is shown in the following image, which shows an alignment of a part of our query sequence (66 residues) with a sequence found in the database.
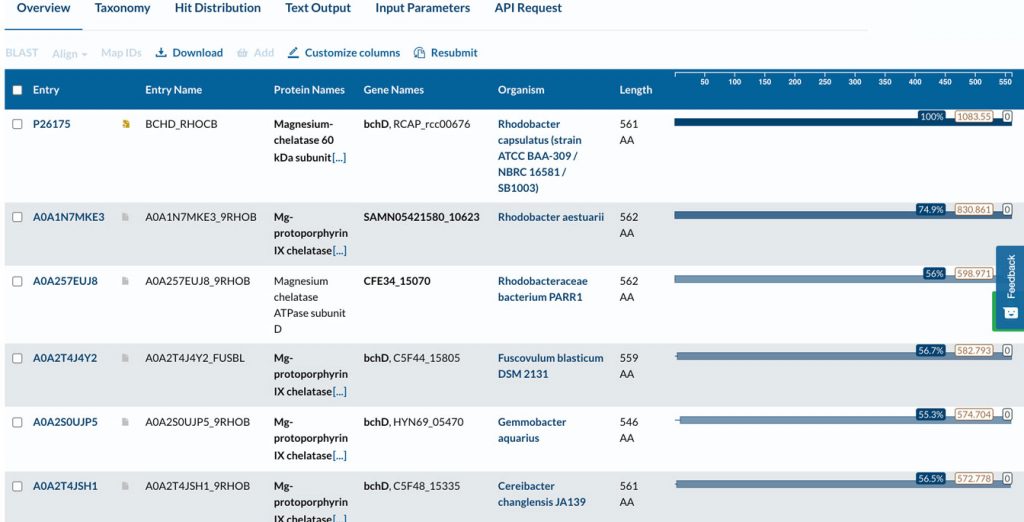

An example of a BLAST search (first image) and a local sequence alignment (residues 1 to 66 included) generated by BLAST at the Uniprot server using the BchI subunit of magnesium chelatase as a search query sequence.
To get deeper into the subject, I recommend the following excellent papers:
Geoffrey J. Barton, Protein sequence alignment and database scanning
Geoffrey J. Barton, Protein Sequence Alignment Techniques
Acta Cryst. (1998). D54, 1139±1146