Example of Structure-Based Drug Design - COX1 & COX2 Specific Inhibition
Introduction
The most straightforward strategy for drug discovery is to identify a biological process/biochemical pathway we want to interfere with, determine the molecular components (i.e., proteins, DNA, RNA, etc.) involved in the pathway and, among those, choose the target for drug design.
As mentioned earlier, the availability of 3D structural information on the complexes of representative compounds with the target substantially accelerates the compound optimization process. Such information becomes crucial when there is, for example, a closely related protein, the inhibition of which is not desirable. For example, kinases are closely related in structure and mechanism of action (about 300 in the human body). Identifying a unique compound that inhibits one particular kinase can be challenging. In other words, a high-affinity and high-specificity binder is required, with the ability to distinguish between closely related proteins. Of course, this process assumes that a biochemical activity assay that can be used to test the compounds has been developed for the target protein.
The drug design example below described a case when structural information was crucial in guiding the design of specific inhibitors targeting two related proteins.
The most straightforward strategy for drug discovery is to identify a biological process/biochemical pathway we want to interfere with, determine the molecular components (i.e., proteins, DNA, RNA, etc.) involved in the pathway and, among those, choose the target for drug design.
As mentioned earlier, the availability of 3D structural information on the complexes of representative compounds with the target substantially accelerates the compound optimization process. Such information becomes crucial when there is, for example, a closely related protein, the inhibition of which is not desirable. For example, kinases are closely related in structure and mechanism of action (about 300 in the human body). Identifying a unique compound that inhibits one particular kinase can be challenging. In other words, a high-affinity and high-specificity binder is required, with the ability to distinguish between closely related proteins. Of course, this process assumes that a biochemical activity assay that can be used to test the compounds has been developed for the target protein.
The drug design example below described a case when structural information was crucial in guiding the design of specific inhibitors targeting two related proteins.

Structural biology services by SARomics Biostructures

X-ray Crystallography

Structure-based drug design

Fragment library screening
Function: COX1, COX2, and prostaglandin signaling
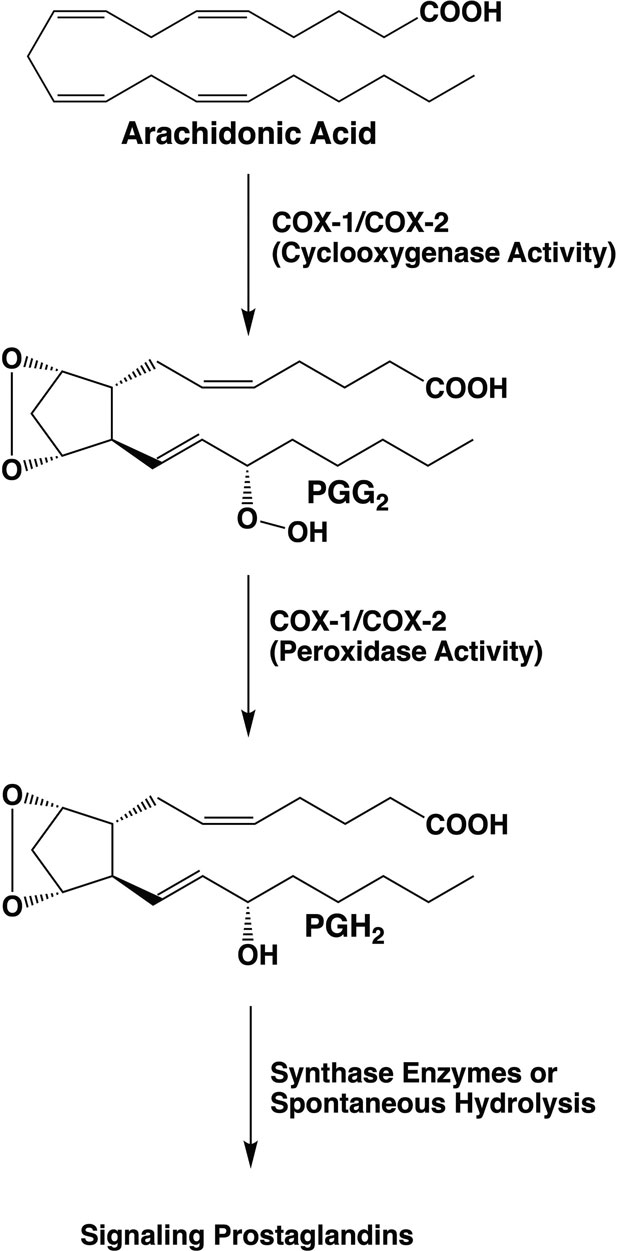
Prostaglandins were discovered in the 1930s by the Swedish biochemist Ulf von Euler. They were called "prostaglandins" because von Euler found them in human semen and thought they were produced in the prostate gland. Further research showed that they were made in almost all tissues of both males and females. Prostaglandins belong to the class of lipid signaling molecules and regulate many cellular processes through the activation of G protein-coupled receptors (GPRC, chemistry Nobel Prize in 2012 to Robert Lefkowitz and Brian Kobilka). The precursor of prostaglandin synthesis in cells is arachidonate, released from membrane phospholipids by the action of phospholipase A2, which is activated by a GPCR receptor. Arachidonate is the substrate of prostaglandin cyclooxygenases 1 and 2 (COX1 and COX2). The COX enzymes play a key role in lipid signaling by catalyzing the first committed step in prostaglandin synthesis. They host two activities, the first is oxygenase, which uses 2 O2 to oxygenate arachidonate, while the second is peroxidase, which removes one of the oxygens (image below).
Prostaglandins were discovered in the 1930s by the Swedish biochemist Ulf von Euler. They were called "prostaglandins" because von Euler found them in human semen and thought they were produced in the prostate gland. Further research showed that they were made in almost all tissues of both males and females. Prostaglandins belong to the class of lipid signaling molecules and regulate many cellular processes through the activation of G protein-coupled receptors (GPRC, chemistry Nobel Prize in 2012 to Robert Lefkowitz and Brian Kobilka). The precursor of prostaglandin synthesis in cells is arachidonate, released from membrane phospholipids by the action of phospholipase A2, which is activated by a GPCR receptor. Arachidonate is the substrate of prostaglandin cyclooxygenases 1 and 2 (COX1 and COX2). The COX enzymes play a key role in lipid signaling by catalyzing the first committed step in prostaglandin synthesis. They host two activities, the first is oxygenase, which uses 2 O2 to oxygenate arachidonate, while the second is peroxidase, which removes one of the oxygens (image below).
COX1 and COX2 share approximately 60% sequence identity and conserved three-dimensional structure. They are also related biochemically and contain cyclooxygenase and peroxidase sites. However, they have different functions in organisms. COX1 is expressed in most tissues and, among other things, produces the prostaglandins required to suppress acid secretion in the stomach and promote the formation of a protective mucus coating of the stomach wall. COX2 expression, on the other hand, is induced by growth factors, inflammatory agents, and tumor promoters. When expressed, COX2 produces prostaglandins that are associated with pain and fever.
Well-known non-steroidal anti-inflammatory drugs (NSAIDs) such as aspirin, paracetamol, and ibuprofen inhibit COX2 and thereby inhibit inflammation-related prostaglandin production. However, these compounds also inhibit COX1, which may result in side effects like gastrointestinal bleeding and ulcer. When this became known, the search started for specific compounds that could selectively inhibit COX2 without inhibiting COX1. The X-ray structures of complexes of COX1 and COX2 with inhibitors provided crucial insights that contributed to a better understanding of the factors that control ligand specificity,
Well-known non-steroidal anti-inflammatory drugs (NSAIDs) such as aspirin, paracetamol, and ibuprofen inhibit COX2 and thereby inhibit inflammation-related prostaglandin production. However, these compounds also inhibit COX1, which may result in side effects like gastrointestinal bleeding and ulcer. When this became known, the search started for specific compounds that could selectively inhibit COX2 without inhibiting COX1. The X-ray structures of complexes of COX1 and COX2 with inhibitors provided crucial insights that contributed to a better understanding of the factors that control ligand specificity,
The cyclooxygenase enzyme reaction
The two-step reaction catalyzed by COX1 and COX2 involves dioxygenation and cyclization of arachidonic acid at the cyclooxygenase active site of COX-1 or COX-2, which yields PGG2. Subsequent reduction of the 15-hydroperoxyl group of PGG2 at the peroxidase active site yields PGH2.
PGH2 serves as a substrate for five different synthases, producing four signaling prostaglandin products (PGE2, PGI2, PGF2α, and PGD2) or thromboxane A2 (TXA2). CA Rouzer & LJ Marnett (2020). Chem. Rev., 120, 15, 7592-7641.
The two-step reaction catalyzed by COX1 and COX2 involves dioxygenation and cyclization of arachidonic acid at the cyclooxygenase active site of COX-1 or COX-2, which yields PGG2. Subsequent reduction of the 15-hydroperoxyl group of PGG2 at the peroxidase active site yields PGH2.
PGH2 serves as a substrate for five different synthases, producing four signaling prostaglandin products (PGE2, PGI2, PGF2α, and PGD2) or thromboxane A2 (TXA2). CA Rouzer & LJ Marnett (2020). Chem. Rev., 120, 15, 7592-7641.
3D structure of COX1 & COX2. Substrate binding
Before starting a drug discovery project, we first need to analyze the target's three-dimensional structure and examine its ligand binding mode. Due to the essential physiological role played by the COX enzyme and the pharmaceutical industry's interest, the structures of many COX1 and COX2 X-ray crystallographic complexes with various ligands (substrate and inhibitors) have been determined. I recommend the excellent review paper discussing the available structures and inhibitors by Rouzer & Marnett (CA Rouzer & LJ Marnett (2020). Chem. Rev., 120, 15, 7592-7641). Here we are primarily interested in the factors that made the design of COX2-specific inhibitors possible.
The close homology of COX1 and COX2 is revealed by analyzing their amino acid sequences. To view the sequence alignment, please click the button below.
The close homology of COX1 and COX2 is revealed by analyzing their amino acid sequences. To view the sequence alignment, please click the button below.
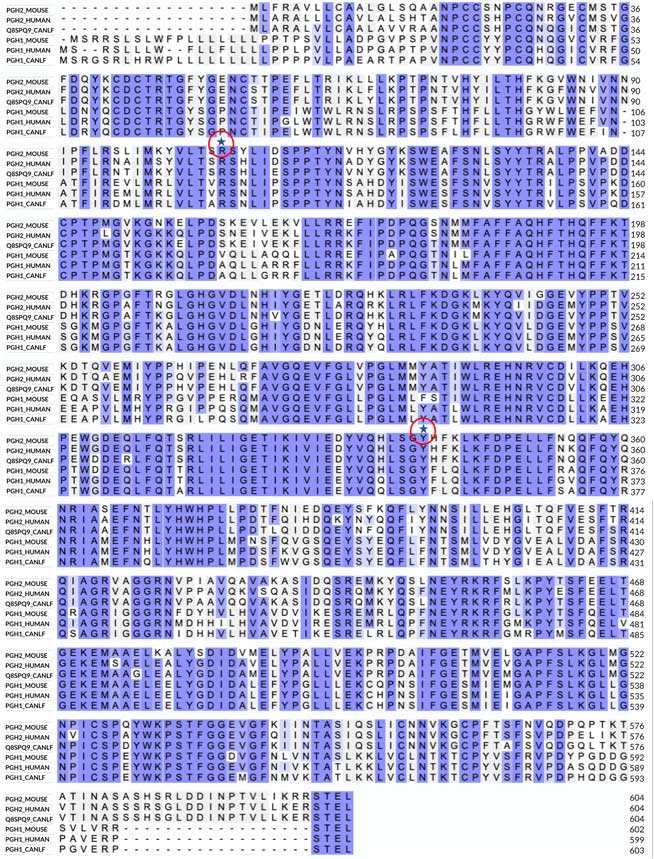
Sequence alignment
In the alignment, the first three sequences are COX2, and the three lower are COX1. The image shows no large insertions or deletions and several highly conserved (invariant) regions in the alignment, which immediately suggests a highly conserved three-dimensional structure. The most notable differences between COX1 and COX2 are the longer N-terminal sequence of COX1 and the longer C-terminal sequence of COX2. I also marked two invariant residues on the alignment, a Tyr and an Arg. As shown below, these residues are essential in binding both substrate and many NSAID inhibitors that contain a carboxyl group.
3D structure
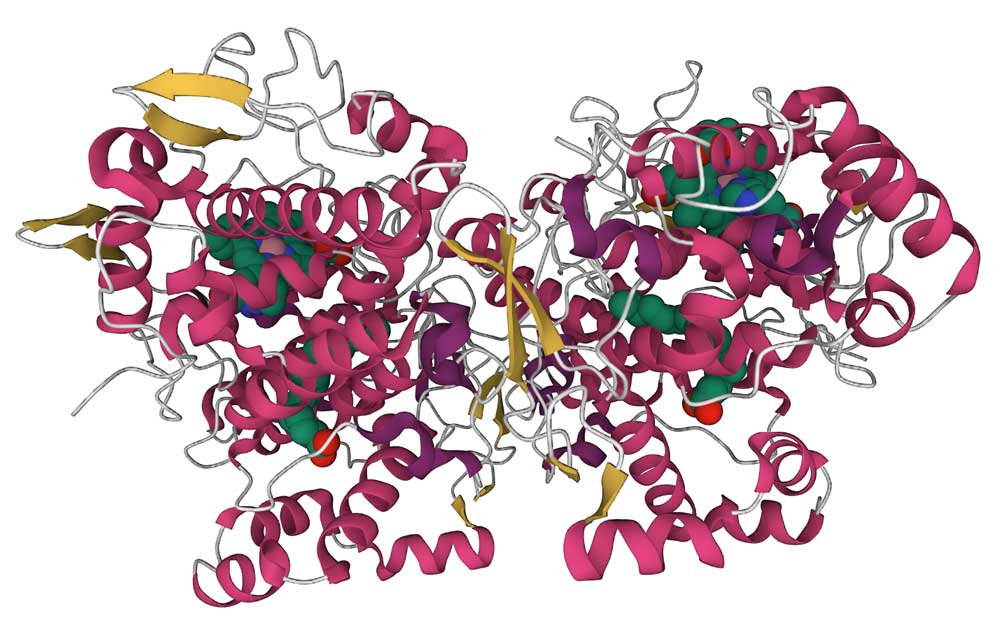
The left image below shows a ribbon representation of the COX1 dimer in a complex with arachidonic acid (PDB ID 1DIY). The CATH database recognizes two domains in each monomer, the small β-hairpin domain (shown in yellow) belongs to the laminin superfamily (CATH Superfamily 2.10.25.10). This fold is found in a large number of proteins with diverse functions. The large helical domain (red color) belongs to the heme peroxidase domain superfamily (CATH Superfamily 1.10.640.10, orthogonal bundle). The smaller domain is located between the two enzyme subunits, presumably stabilizing the interactions between the monomers. The large domain contains the two catalytic sites. It binds the heme prosthetic group (required for both reactions, shown as a space-filling model on the image) between the cyclooxygenase and peroxidase sites. Although, to get a complex with the substrate, the heme group in this structure was replaced by Co-protoporphyrin IX, which does not support the enzymatic reaction. The helices at the bottom of the structure (residues 73-116) run parallel to the lipid membrane and attach the dimer to the lipid bilayer. The sequence alignment shows that this region is among the least conserved. As usual, clicking on the image will take you to the PDB page, where you can look closely at the structure using the graphics program.
Substrate binding
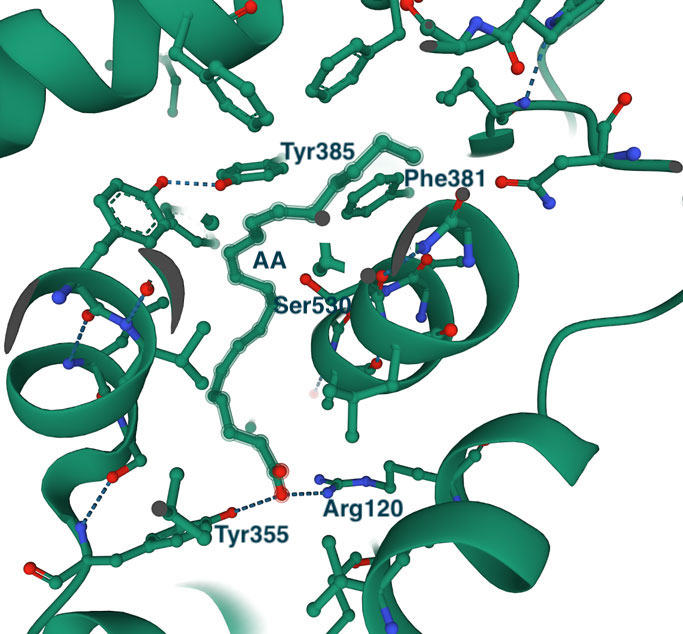
The highly hydrophobic L-shaped substrate binding tunnel (shown as a space-filling model) faces the lipid membrane from which it receives the arachidonic acid (or other substrates of a similar type). The right image shows a close-up of arachidonic acid bound in the active site. I have marked some of the active site residues for orientation. As expected, the binding site is prolonged and highly hydrophobic (the substrate makes 43 hydrophobic contacts with the protein). The invariant Tyr355 and Arg120 at the entrance to the tunnel (also marked on the sequence alignment) interact with the carboxylic group of the substrate or with the polar groups of other ligands, including NSAID inhibitors. The spacious binding site explains the structural diversity of COX substrates and inhibitors. Interesting to note that the binding site of COX2 is about 25% bigger, which, as will be shown below, has implications for the design of COX2-specific inhibitors.
In the middle of the tunnel is the amino acid Ser530. This residue is the target for acetylation by aspirin, one of the most well-known inhibitors of COX enzymes. Acetylation blocks the enzymatic reaction catalyzed by COX1 and COX2, and as you may guess by now, aspirin reacts equally well with both enzymes. Although Ser530 does not participate in the enzymatic reaction, its replacement by a residue with a larger side chain inactivates the enzyme, which suggests that the correct positioning of the substrate at this location is essential for the reaction (reviewed in CA Rouzer & LJ Marnett (2020). Chem. Rev., 120, 15, 7592-7641). Across from Ser530 is Tyr385, a critical residue for the cyclooxygenase reaction. It donates a hydrogen atom to the heme during enzyme activation, which generates a tyrosyl radical that attacks the substrate's hydrogen atom.
In the alignment, the first three sequences are COX2, and the three lower are COX1. The image shows no large insertions or deletions and several highly conserved (invariant) regions in the alignment, which immediately suggests a highly conserved three-dimensional structure. The most notable differences between COX1 and COX2 are the longer N-terminal sequence of COX1 and the longer C-terminal sequence of COX2. I also marked two invariant residues on the alignment, a Tyr and an Arg. As shown below, these residues are essential in binding both substrate and many NSAID inhibitors that contain a carboxyl group.
3D structure
The left image below shows a ribbon representation of the COX1 dimer in a complex with arachidonic acid (PDB ID 1DIY). The CATH database recognizes two domains in each monomer, the small β-hairpin domain (shown in yellow) belongs to the laminin superfamily (CATH Superfamily 2.10.25.10). This fold is found in a large number of proteins with diverse functions. The large helical domain (red color) belongs to the heme peroxidase domain superfamily (CATH Superfamily 1.10.640.10, orthogonal bundle). The smaller domain is located between the two enzyme subunits, presumably stabilizing the interactions between the monomers. The large domain contains the two catalytic sites. It binds the heme prosthetic group (required for both reactions, shown as a space-filling model on the image) between the cyclooxygenase and peroxidase sites. Although, to get a complex with the substrate, the heme group in this structure was replaced by Co-protoporphyrin IX, which does not support the enzymatic reaction. The helices at the bottom of the structure (residues 73-116) run parallel to the lipid membrane and attach the dimer to the lipid bilayer. The sequence alignment shows that this region is among the least conserved. As usual, clicking on the image will take you to the PDB page, where you can look closely at the structure using the graphics program.
Substrate binding
The highly hydrophobic L-shaped substrate binding tunnel (shown as a space-filling model) faces the lipid membrane from which it receives the arachidonic acid (or other substrates of a similar type). The right image shows a close-up of arachidonic acid bound in the active site. I have marked some of the active site residues for orientation. As expected, the binding site is prolonged and highly hydrophobic (the substrate makes 43 hydrophobic contacts with the protein). The invariant Tyr355 and Arg120 at the entrance to the tunnel (also marked on the sequence alignment) interact with the carboxylic group of the substrate or with the polar groups of other ligands, including NSAID inhibitors. The spacious binding site explains the structural diversity of COX substrates and inhibitors. Interesting to note that the binding site of COX2 is about 25% bigger, which, as will be shown below, has implications for the design of COX2-specific inhibitors.
In the middle of the tunnel is the amino acid Ser530. This residue is the target for acetylation by aspirin, one of the most well-known inhibitors of COX enzymes. Acetylation blocks the enzymatic reaction catalyzed by COX1 and COX2, and as you may guess by now, aspirin reacts equally well with both enzymes. Although Ser530 does not participate in the enzymatic reaction, its replacement by a residue with a larger side chain inactivates the enzyme, which suggests that the correct positioning of the substrate at this location is essential for the reaction (reviewed in CA Rouzer & LJ Marnett (2020). Chem. Rev., 120, 15, 7592-7641). Across from Ser530 is Tyr385, a critical residue for the cyclooxygenase reaction. It donates a hydrogen atom to the heme during enzyme activation, which generates a tyrosyl radical that attacks the substrate's hydrogen atom.
COX1 and COX2 inhibitor binding
As mentioned above, the large size of the substrate-binding pocket of the COX enzymes made it possible to design a wide variety of small molecule inhibitors. Structural studies found that most inhibitors occupy the entrance and central part of the substrate tunnel, often called the proximal and central binding pockets of COX1 and COX2. Similarly to the substrate, many inhibitors interact with Tyr355 and Arg120 at the tunnel's entrance.
In addition to the proximal and central pockets, COX2-selective inhibitors were found to occupy the so-called side pocket. The side pocket is created by the replacements of Ile434, Ile523, and His513 of COX1 to Val434, Val523, and Arg513 in COX2, all located in the vicinity of the active site. Ile523Val replacement is probably the most important since it provides access to the side pocket from the substrate binding tunnel.
To illustrate these findings, we will look at the X-ray structures of the complexes of the known anti-inflammatory drug flurbiprofen with COX1 and COX2. After that, we will compare flurbiprofen binding to the complex of the COX2-selective inhibitor SC-558. The complexes of flurbiprofen and SC-558 were published in a Nature paper from 1996 (Kurumbail et al., 1996). Flurbiprofen is a non-selective inhibitor with IC50 values of 0.29 μM and 2.56 μM for COX1 and COX2, respectively.
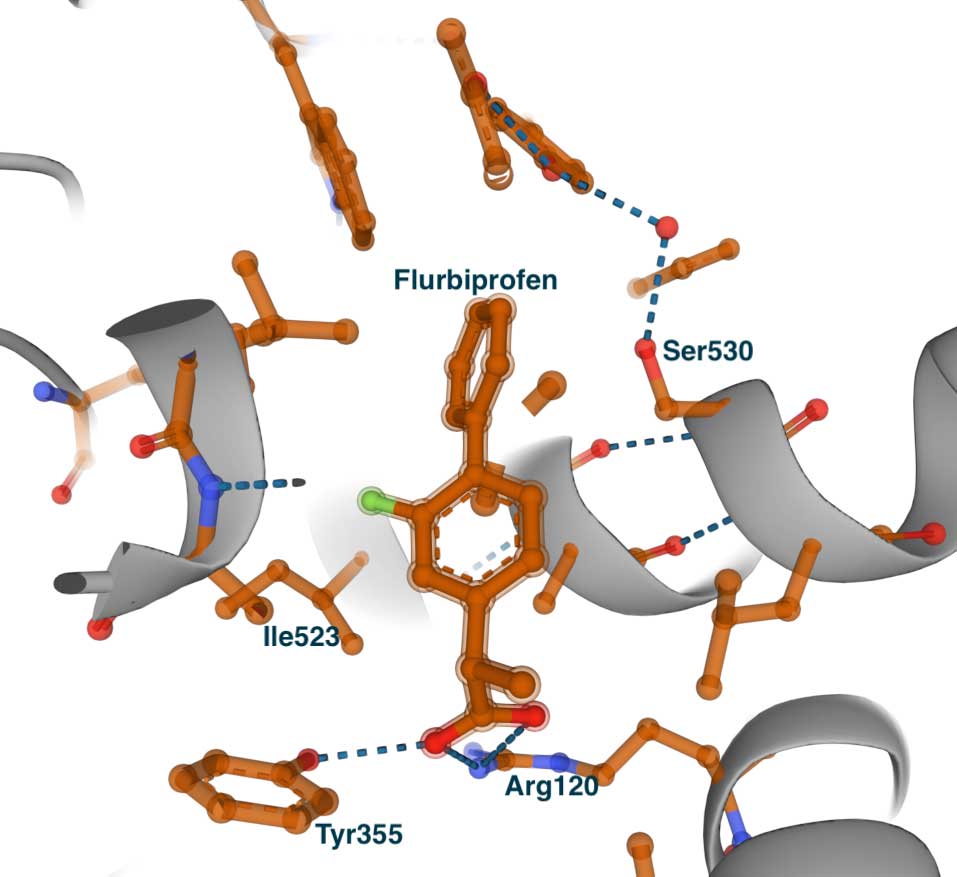
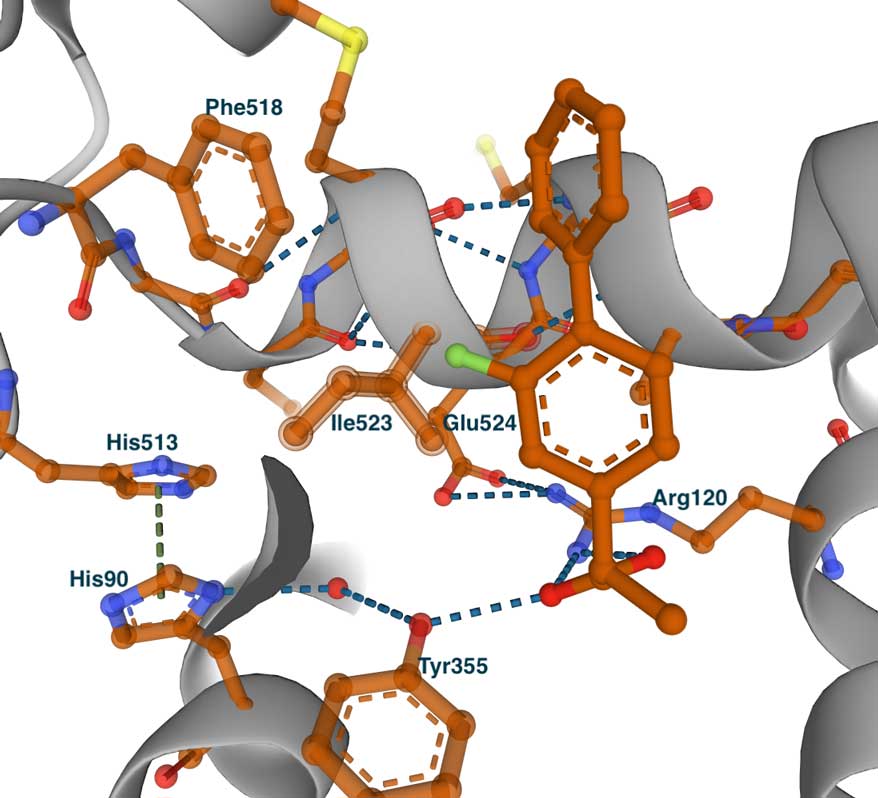
The image on the left shows the complex with COX1 (PDB id 1CQE). The inhibitor binds in the same tunnel as the substrate. It is extended from the entrance, where it makes contact with Tyr355 and Arg120, to the central pocket and Ser530. To demonstrate the concept of the side pocket, on the right is a slightly shifted view of the structure. Some residues that line the side pocket are shown - Phe518, His513, His90, Tyr355, and Ile523. However, we can see that the methyl group of Ile523 blocks the entrance to the pocket. This becomes especially clear when we look at the complex of flurbiprofen with COX2.
In addition to the proximal and central pockets, COX2-selective inhibitors were found to occupy the so-called side pocket. The side pocket is created by the replacements of Ile434, Ile523, and His513 of COX1 to Val434, Val523, and Arg513 in COX2, all located in the vicinity of the active site. Ile523Val replacement is probably the most important since it provides access to the side pocket from the substrate binding tunnel.
To illustrate these findings, we will look at the X-ray structures of the complexes of the known anti-inflammatory drug flurbiprofen with COX1 and COX2. After that, we will compare flurbiprofen binding to the complex of the COX2-selective inhibitor SC-558. The complexes of flurbiprofen and SC-558 were published in a Nature paper from 1996 (Kurumbail et al., 1996). Flurbiprofen is a non-selective inhibitor with IC50 values of 0.29 μM and 2.56 μM for COX1 and COX2, respectively.
The image on the left shows the complex with COX1 (PDB id 1CQE). The inhibitor binds in the same tunnel as the substrate. It is extended from the entrance, where it makes contact with Tyr355 and Arg120, to the central pocket and Ser530. To demonstrate the concept of the side pocket, on the right is a slightly shifted view of the structure. Some residues that line the side pocket are shown - Phe518, His513, His90, Tyr355, and Ile523. However, we can see that the methyl group of Ile523 blocks the entrance to the pocket. This becomes especially clear when we look at the complex of flurbiprofen with COX2.
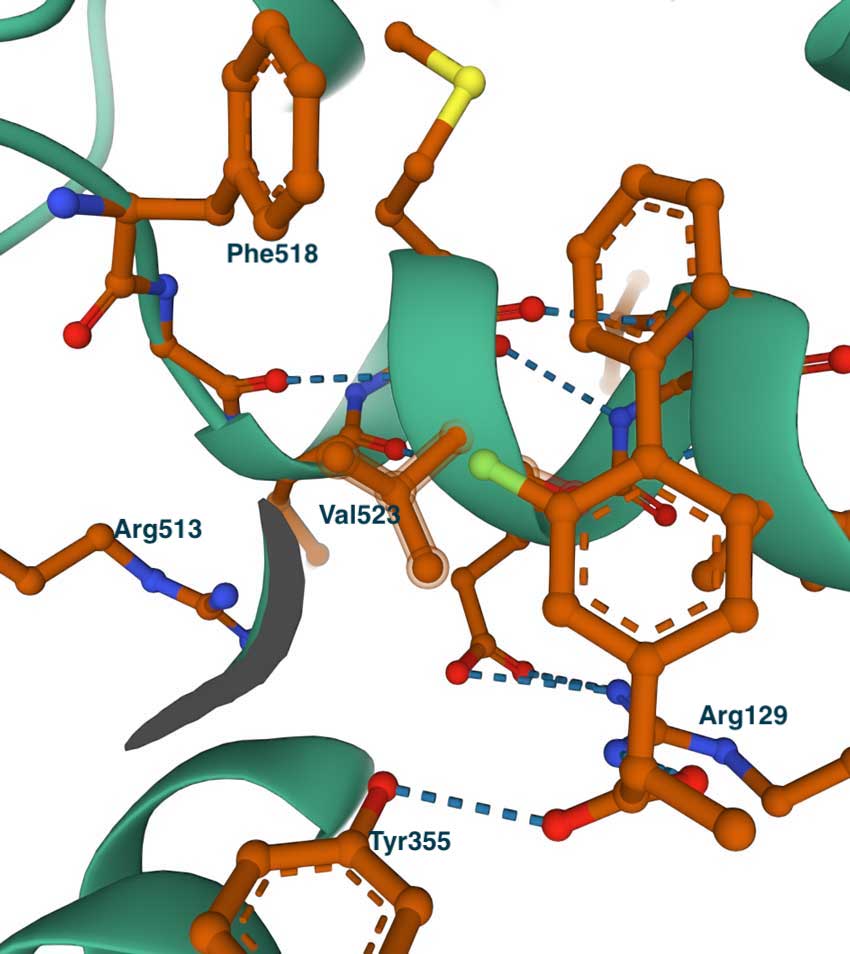
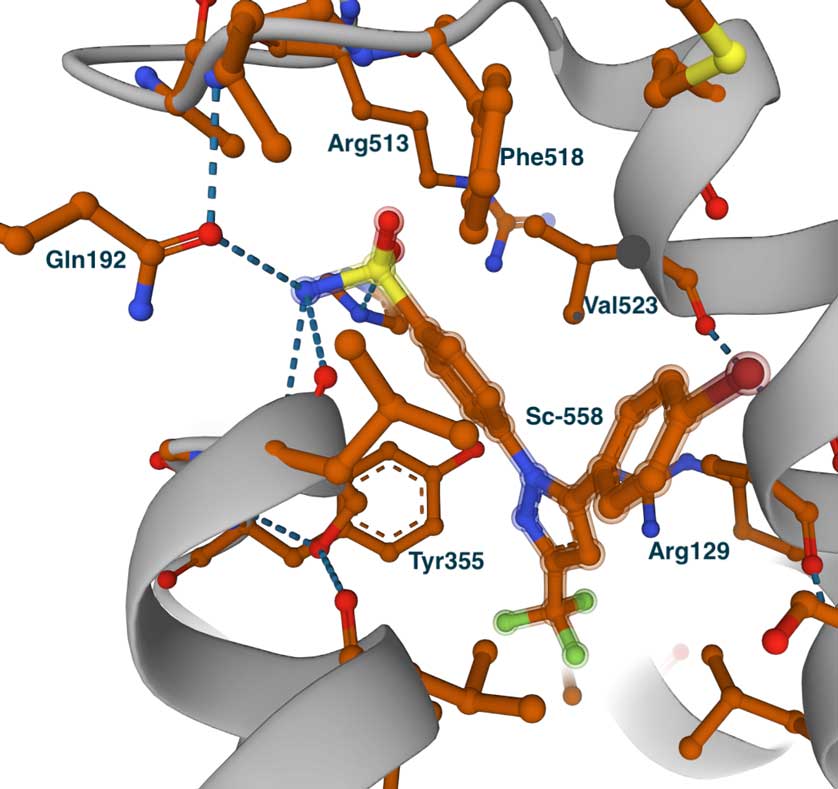
This image on the left shows the complex of COX2 with flurbiprofen (PDB id 3PGH), also showing Val523 and Arg513 (Ile523 and His513 in COX1). Compared with the COX1 complex above, we can notice the wider opening towards the side pocket. Although not shown in this image, the Ile434Val replacement results in a shift of the position of Phe518 and adjacent residues, which widens the pocket and allows the accommodation of larger groups. This can be seen in the image on the right which shows the COX2-specific diarylheterocycle class inhibitor SC-558 complex with COX2 (PDB id 6COX). You can also see the full list of interactions using the LigPlot chart at PDBsum. In this structure, the phenyl sulfonamide group occupies the side pocket of COX2, making hydrogen bonds with Gln192, His90, Ser353, and the main chain carbonyl of Leu252. The other groups of the inhibitor, the pyrazole, and bromophenyl rings occupy the proximal and central binding pockets, respectively. This binding pose is similar to that of other inhibitors like flurbiprofen.
Concluding remarks
This was a short story about the design of COX1 and COX2 inhibitors. The aim was to illustrate how insights gathered from the three-dimensional structure can help in the design of specific inhibitors. The take-home lesson is that we always need to know the details of the function of the protein we are targeting. We need careful studies of enzyme kinetics and detailed ligand binding studies, often combined with mutations, to reveal the role of the different active site residues, and of course, we need structures of ligand complexes with the protein. A more detailed story of COX2-COX2 can be found in the review I cited earlier, CA Rouzer & LJ Marnett (2020). Chem. Rev., 120, 15, 7592-7641.
This was a short story about the design of COX1 and COX2 inhibitors. The aim was to illustrate how insights gathered from the three-dimensional structure can help in the design of specific inhibitors. The take-home lesson is that we always need to know the details of the function of the protein we are targeting. We need careful studies of enzyme kinetics and detailed ligand binding studies, often combined with mutations, to reveal the role of the different active site residues, and of course, we need structures of ligand complexes with the protein. A more detailed story of COX2-COX2 can be found in the review I cited earlier, CA Rouzer & LJ Marnett (2020). Chem. Rev., 120, 15, 7592-7641.