Lead Discovery, Lead Optimization & Structure-Based Drug Design: Overview
Before starting a drug discovery project, we need to identify a target. Most small-molecule drugs target protein molecules. Although nucleic acids may also be considered, their use as drug targets in drug discovery and structure-based drug design (SBDD) has been limited due to various reasons like toxicity, difficulty in achieving high specificity, etc. There is also a large class of biological drugs, most of which are antibodies. Here we are going to focus on small-molecule drugs.
For illustration, a survey of marketed drugs published in 2006 showed that there were 1357 unique drugs. Of those, 1204 were small-molecule drugs, while 166 were biological agents (mainly antibodies). The total number of drug targets for human and pathogenic organisms was 324 (Overington, Al-Lazikani & Hopkins, 2006). A more recent study by the same group from 2016 showed a substantial increase in the number of drug targets to 893 (667 human-derived targets), while the number of FDA-approved drugs was 1578, of which 195 were biologics (Santos et al., 2016). This shows that most marketable drugs are still small-molecule drugs, although the number (but not proportion!) of biologics is growing. I should mention that there are different ways to make these calculations, and different authors may arrive at other numbers; however, the studies cited here were performed by the same group of authors, which makes the comparison more reliable.
The questions we can ask now: How can structural biology and structural bioinformatics help us in lead discovery & lead optimization, a process generally called structure-based drug design? To answer these questions, we first need to look at the different stages of the drug discovery process to identify areas where structural biology may be applied. The image below provides an integrated view of the various stages of the drug discovery process.
For illustration, a survey of marketed drugs published in 2006 showed that there were 1357 unique drugs. Of those, 1204 were small-molecule drugs, while 166 were biological agents (mainly antibodies). The total number of drug targets for human and pathogenic organisms was 324 (Overington, Al-Lazikani & Hopkins, 2006). A more recent study by the same group from 2016 showed a substantial increase in the number of drug targets to 893 (667 human-derived targets), while the number of FDA-approved drugs was 1578, of which 195 were biologics (Santos et al., 2016). This shows that most marketable drugs are still small-molecule drugs, although the number (but not proportion!) of biologics is growing. I should mention that there are different ways to make these calculations, and different authors may arrive at other numbers; however, the studies cited here were performed by the same group of authors, which makes the comparison more reliable.
The questions we can ask now: How can structural biology and structural bioinformatics help us in lead discovery & lead optimization, a process generally called structure-based drug design? To answer these questions, we first need to look at the different stages of the drug discovery process to identify areas where structural biology may be applied. The image below provides an integrated view of the various stages of the drug discovery process.
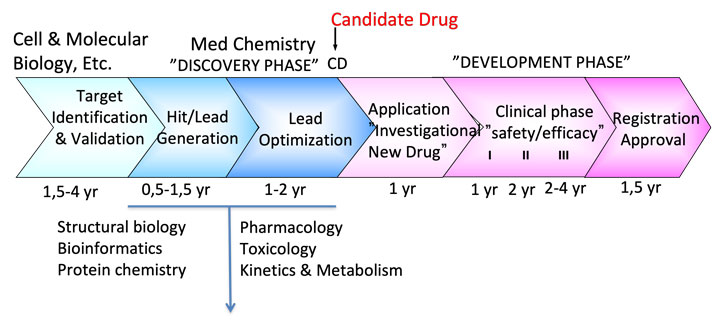
The image indicates that the first three stages of the drug discovery process require structural information.
1) Target identification & validation - three-dimensional protein structure is required in studying the protein in question, e.g., the location of the substrate binding site (or sites), its reaction mechanism, evolutionary relationships, etc.
2) Hit identification/lead generation - together with computational chemistry, medicinal chemistry, biochemistry, etc., structural information will contribute to a considerable acceleration of the drug discovery process.
3) Lead optimization - at this stage, the protein structure will significantly help map the interactions made by the tested compounds and plan subsequent optimizations of compound parameters.
It is also clear from the image that the drug discovery process is highly multidisciplinary and requires expertise in a broad range of sciences.
Planning a structure-based drug design project
Before starting a structure-based lead discovery project, some general considerations must be made. We assume that the protein target has been identified and verified. Since we aim to identify a small molecule compound that will bind to the protein and somehow modulate its activity, the protein should be well studied in the lab. For example, if it is an enzyme, it would be a huge advantage to have a good idea about the type and mechanism of the biochemical reaction catalyzed, the availability of inhibitors, co-factor requirement, the need for an allosteric regulator, the presence of metals, etc.
For structure-based drug design, a high resolution three-dimensional structure of the protein is a requirement, either from Protein crystallography, NMR spectroscopy, or cry-electron microscopy. The resolution of the structure needs to be at least better than 3 Å, but preferably 2.5 Å and better. As mentioned earlier, the higher the resolution, the more accurate the structure will be, and our description of ligand-protein interactions will be more precise.
Generally, the drug design process is repetitive. The hits initially identified through screening against a small-molecule compound library will generally need to be optimized in several cycles until we have a so-called lead molecule (a process called lead discovery & lead optimization). If a fragment library is used for screening, after hit identification, we will enter a process called hit expansion, described later in the chapter. The aim is to identify drug-like molecules with stable binding, good solubility, and low toxicity that would not be very expensive to synthesize.
In a case when known inhibitors of the enzyme already exist (an excellent place to look for those can be the PDBe Chemical Components Library), the structures of the inhibitors in complex with the protein can be used for mapping the interactions within the binding site and for build a so-called pharmacophore model for the binding site. Such models can be used, among other things, in filtering compound libraries before screening for potential binders. There is no meaning in including in the library compounds that would not fit into the protein binding site, for example, due to bad shape complementarity, absence of groups that would interact with the binding-site amino acids, etc.
Suppose there is no experimental three-dimensional structure available for the protein target. In that case, one could try to find a predicted structure of the protein in the AlphaFold database, which may be used at the initial stage of the project.
We should remember that the structural part, even if it is essential, is a relatively small and cheap part of a drug discovery project. The main ideas presented on this page are discussed in more detail on the following pages, including an example of how structural insights helped design specific inhibitors for the cyclooxygenase 2 (COX2) enzyme.
1) Target identification & validation - three-dimensional protein structure is required in studying the protein in question, e.g., the location of the substrate binding site (or sites), its reaction mechanism, evolutionary relationships, etc.
2) Hit identification/lead generation - together with computational chemistry, medicinal chemistry, biochemistry, etc., structural information will contribute to a considerable acceleration of the drug discovery process.
3) Lead optimization - at this stage, the protein structure will significantly help map the interactions made by the tested compounds and plan subsequent optimizations of compound parameters.
It is also clear from the image that the drug discovery process is highly multidisciplinary and requires expertise in a broad range of sciences.
Planning a structure-based drug design project
Before starting a structure-based lead discovery project, some general considerations must be made. We assume that the protein target has been identified and verified. Since we aim to identify a small molecule compound that will bind to the protein and somehow modulate its activity, the protein should be well studied in the lab. For example, if it is an enzyme, it would be a huge advantage to have a good idea about the type and mechanism of the biochemical reaction catalyzed, the availability of inhibitors, co-factor requirement, the need for an allosteric regulator, the presence of metals, etc.
For structure-based drug design, a high resolution three-dimensional structure of the protein is a requirement, either from Protein crystallography, NMR spectroscopy, or cry-electron microscopy. The resolution of the structure needs to be at least better than 3 Å, but preferably 2.5 Å and better. As mentioned earlier, the higher the resolution, the more accurate the structure will be, and our description of ligand-protein interactions will be more precise.
Generally, the drug design process is repetitive. The hits initially identified through screening against a small-molecule compound library will generally need to be optimized in several cycles until we have a so-called lead molecule (a process called lead discovery & lead optimization). If a fragment library is used for screening, after hit identification, we will enter a process called hit expansion, described later in the chapter. The aim is to identify drug-like molecules with stable binding, good solubility, and low toxicity that would not be very expensive to synthesize.
In a case when known inhibitors of the enzyme already exist (an excellent place to look for those can be the PDBe Chemical Components Library), the structures of the inhibitors in complex with the protein can be used for mapping the interactions within the binding site and for build a so-called pharmacophore model for the binding site. Such models can be used, among other things, in filtering compound libraries before screening for potential binders. There is no meaning in including in the library compounds that would not fit into the protein binding site, for example, due to bad shape complementarity, absence of groups that would interact with the binding-site amino acids, etc.
Suppose there is no experimental three-dimensional structure available for the protein target. In that case, one could try to find a predicted structure of the protein in the AlphaFold database, which may be used at the initial stage of the project.
We should remember that the structural part, even if it is essential, is a relatively small and cheap part of a drug discovery project. The main ideas presented on this page are discussed in more detail on the following pages, including an example of how structural insights helped design specific inhibitors for the cyclooxygenase 2 (COX2) enzyme.

Structural biology services by SARomics Biostructures

X-ray Crystallography

NMR spectroscopy

Structure-based drug design

Fragment library screening