Sequence Alignment and Analysis Tutorial
Background
Amino acid sequence alignment is a great tool in the study of proteins. Nowadays, we can find much information about the protein using only the sequence. And with the arrival of AlphaFold and the ESM Metagenomic Atlas, even the predicted three-dimensional structure is now available for our analysis. Still, alignment of the amino acid sequence may provide valuable insights into the protein's conservation pattern, the presence of functionally important conserved motifs, domain structure, evolutionary relationships within the family, etc.
This tutorial will analyze two related proteins, two subunits of the enzyme magnesium chelatase, BchI, and BchD. I will focus on using the tools available at UniProt and EBI multiple sequence alignment (MSA) servers. Although many other servers will do the same job, I am used to these two. We will explore how to make a sequence alignment. At the same time, we will examine the information in UniProt and other related databases to learn more about these two proteins.
Subunits BchI (35 kDa) and BchD (70 kDa) together build up a large 600 kDa complex (see an image showing the cryo-EM reconstruction of the complex); there is also a third subunit required for the enzyme's function called BchH (about 120 kDa). However, we are not going to discuss it here. Magnesium chelatase catalyzes the insertion of an Mg2+ ion into protoporphyrin IX at the first committed step of chlorophyll biosynthesis. As we can guess from the size, BchI and BchD are multidomain proteins. In such cases, we need to define the domains and, by analyzing any conservation patterns, try to identify possible relationships between them and other proteins. Insights gathered from the such analysis may help us better understand the mechanism of function of the enzyme. There are, of course, different ways of running such analysis. I will be showing just one which I think is logical. After learning how to use the tools described here, you will be able to design your analysis routines, whatever the aim of your analysis may be.
Amino acid sequence alignment is a great tool in the study of proteins. Nowadays, we can find much information about the protein using only the sequence. And with the arrival of AlphaFold and the ESM Metagenomic Atlas, even the predicted three-dimensional structure is now available for our analysis. Still, alignment of the amino acid sequence may provide valuable insights into the protein's conservation pattern, the presence of functionally important conserved motifs, domain structure, evolutionary relationships within the family, etc.
This tutorial will analyze two related proteins, two subunits of the enzyme magnesium chelatase, BchI, and BchD. I will focus on using the tools available at UniProt and EBI multiple sequence alignment (MSA) servers. Although many other servers will do the same job, I am used to these two. We will explore how to make a sequence alignment. At the same time, we will examine the information in UniProt and other related databases to learn more about these two proteins.
Subunits BchI (35 kDa) and BchD (70 kDa) together build up a large 600 kDa complex (see an image showing the cryo-EM reconstruction of the complex); there is also a third subunit required for the enzyme's function called BchH (about 120 kDa). However, we are not going to discuss it here. Magnesium chelatase catalyzes the insertion of an Mg2+ ion into protoporphyrin IX at the first committed step of chlorophyll biosynthesis. As we can guess from the size, BchI and BchD are multidomain proteins. In such cases, we need to define the domains and, by analyzing any conservation patterns, try to identify possible relationships between them and other proteins. Insights gathered from the such analysis may help us better understand the mechanism of function of the enzyme. There are, of course, different ways of running such analysis. I will be showing just one which I think is logical. After learning how to use the tools described here, you will be able to design your analysis routines, whatever the aim of your analysis may be.
Characteristic features of a quence. Running Blast
In the beginning, we need to choose the sequences for the alignment. To start, we write the name of the protein (BchD) into the UniProt search window, and we will be taken to a list of sequences of the protein from different organisms.
The site shows a large number of sequences. You may notice on the left, where it says “Filtered by,” that there are “Reviewed” and “Unreviewed” sequences. This is one of my favorite features - when there is a sufficient number of reviewed sequences, I usually choose them for further analysis. These sequences have been verified to be what we expect them to be. There are many automatically annotated sequences among the Unreviewed, and sometimes we may find assignment errors. There is more helpful information (on the left field), like the availability of a 3D structure, catalytic activity, etc.
We will choose BCHD_RHOCB (entry P26175), the subunit BchD from Rhodobacter capsulatus. In plants, the homologous subunit is called ChlD; "B" in BchD indicates that the protein is involved in bacteriochlorophyll syntheses, while ChlD is related to chlorophyll synthesis.
On the page that will open after clicking the entry ID P26175, we will find a lot of detailed information on magnesium chelatase - its biological function (photosynthesis, magnesium chelatase activity), type of ligands/substrate it binds, catalytic function (insertion of magnesium and ATP hydrolysis), links to published works, links to entries related to this particular protein in other databases, and of course the amino acid sequence of BchD.
Clicking "Family & Domains" on the menu on the left of the UniProt page will show the domain content of BchD. However, in this case, we only get the vWFA domain (von Willebrand Factor A-like domain superfamily) at the C-terminal of the protein (residues 375-559). The InterPro database link also directs us to the BchD/ChlD, vWFA domain page. It is nice to see that they reference our publication from 2001, Fodje et al., J. Mol. Biol. 311, 111-22. We were the first to identify this domain in the magnesium chelatase family. A characteristic feature of the vWFA domain is the conservation of the so-called MIDAS motif. MIDAS stands for metal ion-dependent adhesion site (see a paper by Lacy et al. 2004 for a detailed description of the structure). The conserved sequence consists of the DXSXS motif and additional threonine and aspartate (T and D) residues. The DXSXS motif residues in BchD are D385, S387 S389, which are close to the N-terminus of the vWFA domain, while threonine T452 and aspartate D482 are located further down in the sequence. The alignment in the image below (click to see it, it can also be found at the Conserved Protein Domain Family server at NCBI) shows the position of these invariant residues (yellow boxes, also marked by a # on top of the alignment). These residues are involved in metal ion binding, Ca2+ or Mg2+. We still do not know the exact function of this domain within the magnesium chelatase complex.
The site shows a large number of sequences. You may notice on the left, where it says “Filtered by,” that there are “Reviewed” and “Unreviewed” sequences. This is one of my favorite features - when there is a sufficient number of reviewed sequences, I usually choose them for further analysis. These sequences have been verified to be what we expect them to be. There are many automatically annotated sequences among the Unreviewed, and sometimes we may find assignment errors. There is more helpful information (on the left field), like the availability of a 3D structure, catalytic activity, etc.
We will choose BCHD_RHOCB (entry P26175), the subunit BchD from Rhodobacter capsulatus. In plants, the homologous subunit is called ChlD; "B" in BchD indicates that the protein is involved in bacteriochlorophyll syntheses, while ChlD is related to chlorophyll synthesis.
On the page that will open after clicking the entry ID P26175, we will find a lot of detailed information on magnesium chelatase - its biological function (photosynthesis, magnesium chelatase activity), type of ligands/substrate it binds, catalytic function (insertion of magnesium and ATP hydrolysis), links to published works, links to entries related to this particular protein in other databases, and of course the amino acid sequence of BchD.
Clicking "Family & Domains" on the menu on the left of the UniProt page will show the domain content of BchD. However, in this case, we only get the vWFA domain (von Willebrand Factor A-like domain superfamily) at the C-terminal of the protein (residues 375-559). The InterPro database link also directs us to the BchD/ChlD, vWFA domain page. It is nice to see that they reference our publication from 2001, Fodje et al., J. Mol. Biol. 311, 111-22. We were the first to identify this domain in the magnesium chelatase family. A characteristic feature of the vWFA domain is the conservation of the so-called MIDAS motif. MIDAS stands for metal ion-dependent adhesion site (see a paper by Lacy et al. 2004 for a detailed description of the structure). The conserved sequence consists of the DXSXS motif and additional threonine and aspartate (T and D) residues. The DXSXS motif residues in BchD are D385, S387 S389, which are close to the N-terminus of the vWFA domain, while threonine T452 and aspartate D482 are located further down in the sequence. The alignment in the image below (click to see it, it can also be found at the Conserved Protein Domain Family server at NCBI) shows the position of these invariant residues (yellow boxes, also marked by a # on top of the alignment). These residues are involved in metal ion binding, Ca2+ or Mg2+. We still do not know the exact function of this domain within the magnesium chelatase complex.

It is a bit disappointing that they only mention the vWFA domain and don't mention the homology of the N-terminal sequence of BchD to the BchI subunit sequence. However, there is a hint on that; another InterPro link refers to the page on "P-loop containing nucleoside triphosphate hydrolase," and there they mention ChlI and ChlD. Probably the reason is that BchD, in contrast to BchI and ChlI, lacks ATPase activity. The characteristic P-loop sequence motif, which is [AG]-x(4)-G-K-[ST] according to the Prosite database, is not conserved in R. capsulatus BchD; we will look closer into this when we analyze the sequence alignment.
Moving closer to the bottom of the UniProt page we will find the sequence of the protein:
Moving closer to the bottom of the UniProt page we will find the sequence of the protein:
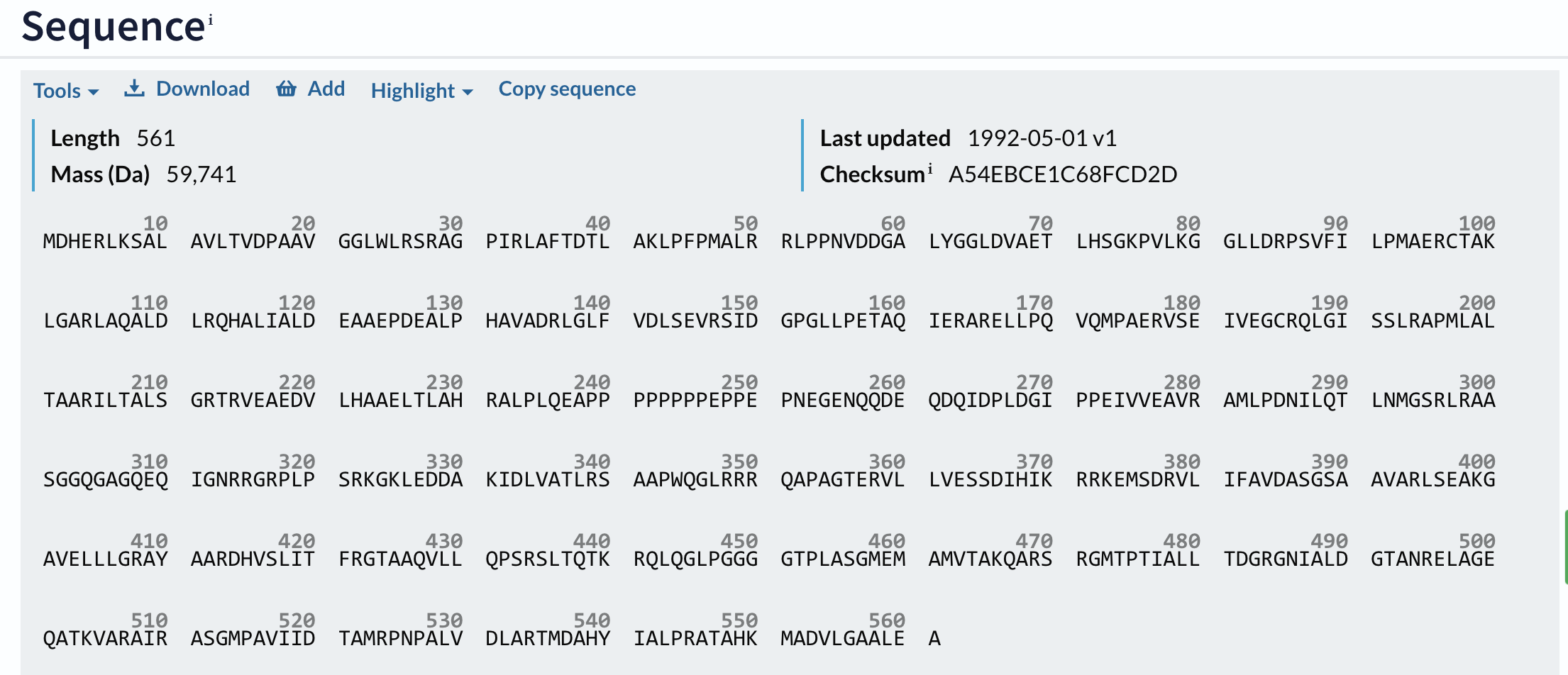
Since we need several sequences for the multiple sequence alignment, I usually run BLAst (Basic Local Alignment, briefly discussed in the introduction) to get a list of related sequences. To run Blast, we can choose "Tools" (shown in the image above in the left corner) and there click "Blast." On the Blast page, you may also choose the substitution matrix you want to use in the search. Blast searches the entire database and outputs a list of sequences aligned against our query sequence (pairwise alignment). I choose several sequences from the list according to organism and percentage sequence identity and save them in the Basket.
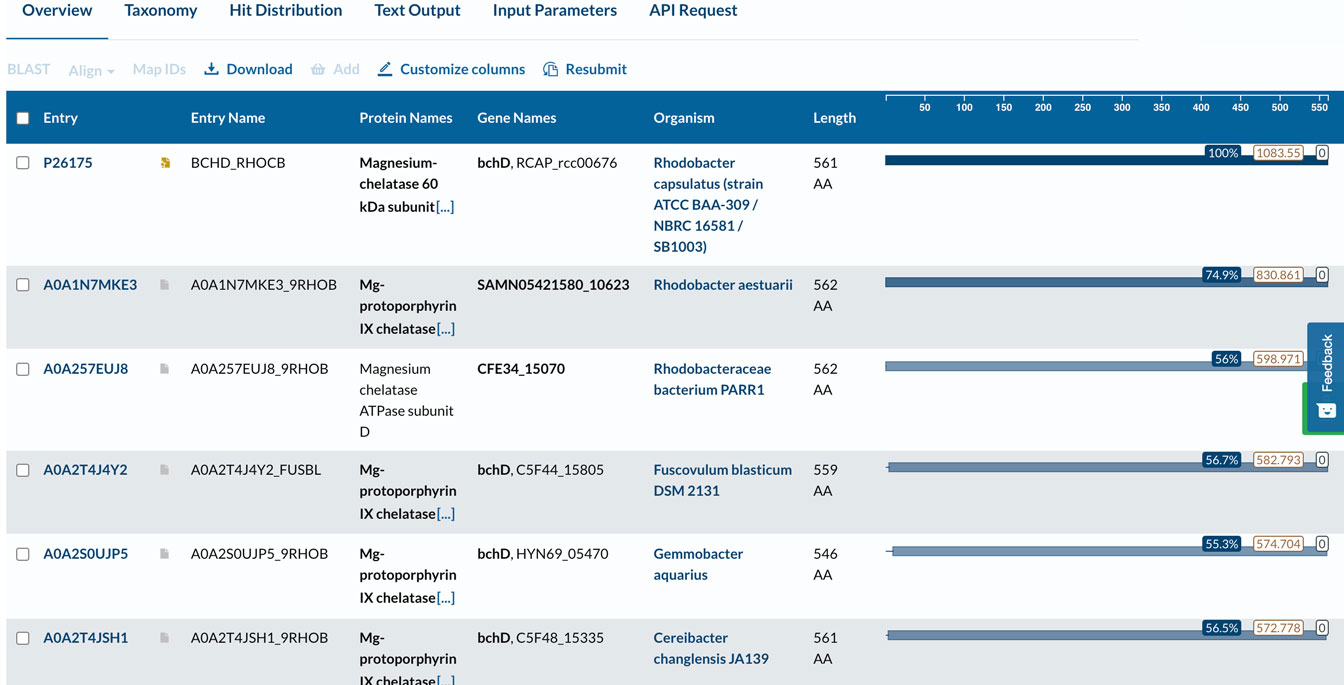
Click below to see part of the list after running Blast. In the image, we can see the sequence identity with the individual organism, and clicking there will show the alignment of the query sequence to the protein found in the database (figure below).
Click below to see part of the list after running Blast. In the image, we can see the sequence identity with the individual organism, and clicking there will show the alignment of the query sequence to the protein found in the database (figure below).

Making the alignment
For the alignment, a good strategy is to choose 3-4 sequences since this will give a better idea of the conservation pattern in the protein family. To run the alignment, I mark the sequences I saved in the Basket and choose "Align" from the Basket menu. We will be taken to the alignment page. A helpful option is related to the order of sequences in the alignment output (Output sequence order) under "Advance parameters." I prefer to use the "input order" to group bacterial and plant sequences separately. This will simplify the analysis of the conservation pattern. We can also edit the sequences in the alignment window if we need.
In the alignment window, the sequences are in the FASTA format, which is different from the format shown as default on the UniProt page. If we need the sequence in FASTA format, we need to click "Download" above the sequence shown in UniProt, then save it by copying and pasting it for later use or insert it into the MSA server alignment window if we choose to run the alignment there. At the bottom of this page, you will find an explanation of the FASTA format.
I usually remove most of the information on top of the sequence (everything which comes after the "larger than" sign > and only leave the name of the protein, e.g., BCHD_RHOCB. This will simplify the analysis of the alignment. Don’t forget to include the protein from Rhodobacter capsulatus.
Click below to see the alignment which I made.
For the alignment, a good strategy is to choose 3-4 sequences since this will give a better idea of the conservation pattern in the protein family. To run the alignment, I mark the sequences I saved in the Basket and choose "Align" from the Basket menu. We will be taken to the alignment page. A helpful option is related to the order of sequences in the alignment output (Output sequence order) under "Advance parameters." I prefer to use the "input order" to group bacterial and plant sequences separately. This will simplify the analysis of the conservation pattern. We can also edit the sequences in the alignment window if we need.
In the alignment window, the sequences are in the FASTA format, which is different from the format shown as default on the UniProt page. If we need the sequence in FASTA format, we need to click "Download" above the sequence shown in UniProt, then save it by copying and pasting it for later use or insert it into the MSA server alignment window if we choose to run the alignment there. At the bottom of this page, you will find an explanation of the FASTA format.
I usually remove most of the information on top of the sequence (everything which comes after the "larger than" sign > and only leave the name of the protein, e.g., BCHD_RHOCB. This will simplify the analysis of the alignment. Don’t forget to include the protein from Rhodobacter capsulatus.
Click below to see the alignment which I made.
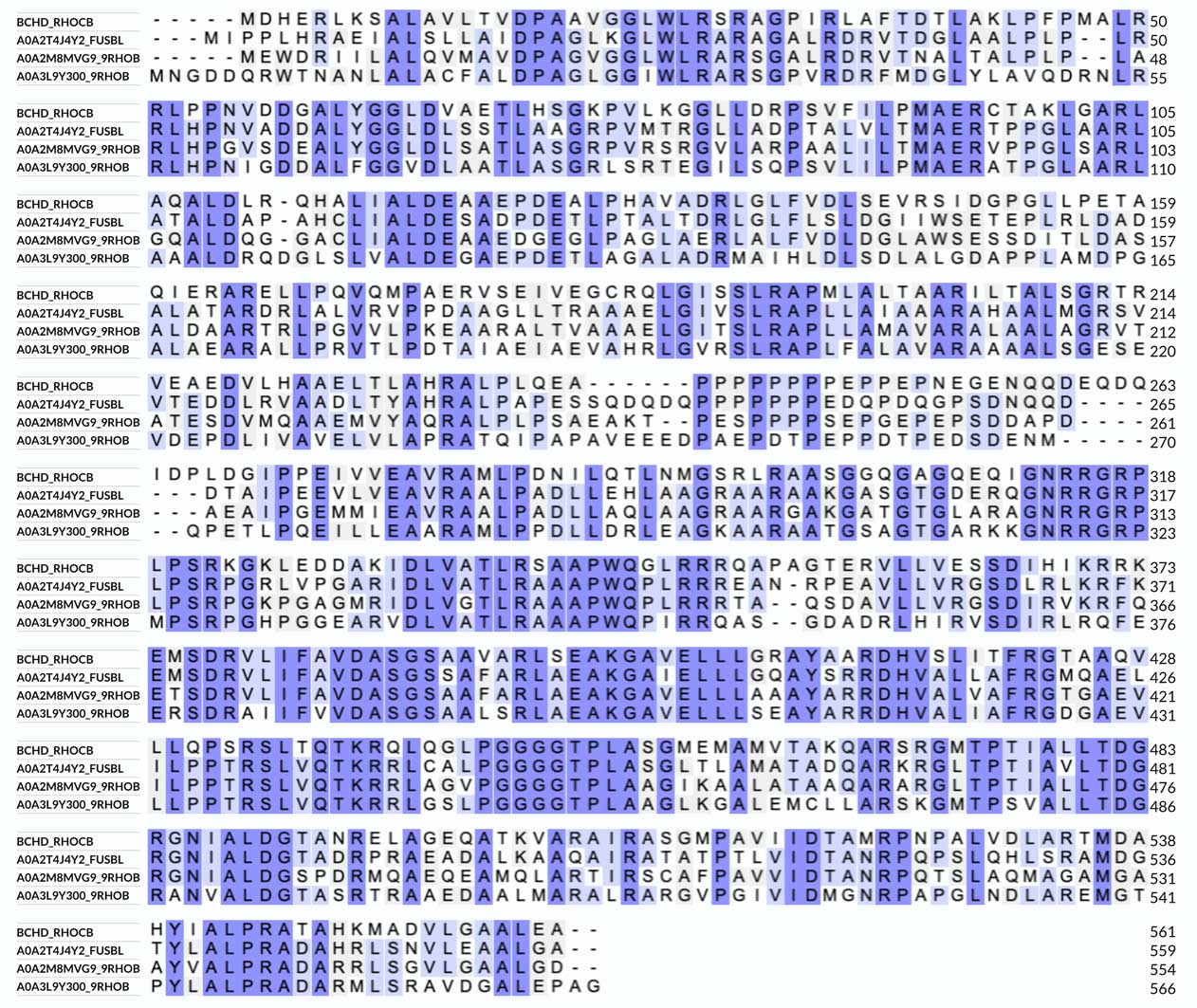
Analyzing the alignment
The alignment is colored according to similarity. There are other options for coloring, e.g., by residue type, which will show hydrophobic, charged, and polar amino acids. We can see that the sequence of BchD is well-conserved among different bacteria - there are just a few insertions and deletions.
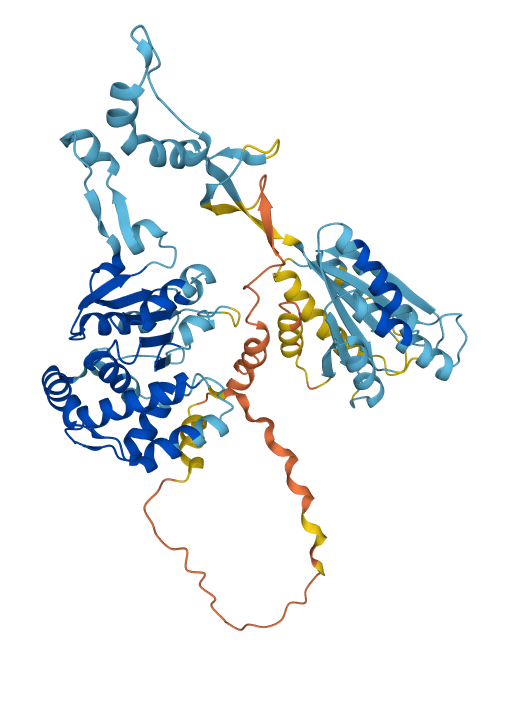
We can also examine the AlphaFold predicted structure of BchD (image below) to get additional insights into the sequence-structure relationships for this protein. As mentioned above, the C-terminal domain (residues 375-559) was earlier recognized as a vWAF-type domain. The predicted structure shows that a long stretch of the sequence that does not seem to have much secondary structure connects the C-terminal domain to the N-terminal domain. The residues from approximately 232 to 309 (the orange color means prediction is unreliable) are followed by a region with some secondary structure which continues until Met375, where the vWAF domain starts. Looking at the alignment, we can see that up to Glu309 we have a so-called low-complexity region - it is not well conserved, and there are many repeats but also many acidic residues. Such regions are usually very flexible and often found to be involved in protein-protein interactions. This suggests that this region may somehow interact with other subunits within the large 600 kDa magnesium chelatase complex. The rest of the sequence, up to the vWAF domain, appears to be more structures, although judging by the color, the reliability of the prediction is still low there. The N-terminal domain, which contains residues 1-232, as demonstrated in the above-mentioned publication by Fodje et al., has a conserved fold of the so-called AAA+ family of proteins to which the smallest magnesium chelatase subunit BchI belongs. It is possible to verify this by submitting the AlphaFold predicted structure to one of the fold recognition servers like DALI. We can also verify the homology between this domain by an alignment of the sequences of BchD and BchI.
The alignment is colored according to similarity. There are other options for coloring, e.g., by residue type, which will show hydrophobic, charged, and polar amino acids. We can see that the sequence of BchD is well-conserved among different bacteria - there are just a few insertions and deletions.
We can also examine the AlphaFold predicted structure of BchD (image below) to get additional insights into the sequence-structure relationships for this protein. As mentioned above, the C-terminal domain (residues 375-559) was earlier recognized as a vWAF-type domain. The predicted structure shows that a long stretch of the sequence that does not seem to have much secondary structure connects the C-terminal domain to the N-terminal domain. The residues from approximately 232 to 309 (the orange color means prediction is unreliable) are followed by a region with some secondary structure which continues until Met375, where the vWAF domain starts. Looking at the alignment, we can see that up to Glu309 we have a so-called low-complexity region - it is not well conserved, and there are many repeats but also many acidic residues. Such regions are usually very flexible and often found to be involved in protein-protein interactions. This suggests that this region may somehow interact with other subunits within the large 600 kDa magnesium chelatase complex. The rest of the sequence, up to the vWAF domain, appears to be more structures, although judging by the color, the reliability of the prediction is still low there. The N-terminal domain, which contains residues 1-232, as demonstrated in the above-mentioned publication by Fodje et al., has a conserved fold of the so-called AAA+ family of proteins to which the smallest magnesium chelatase subunit BchI belongs. It is possible to verify this by submitting the AlphaFold predicted structure to one of the fold recognition servers like DALI. We can also verify the homology between this domain by an alignment of the sequences of BchD and BchI.
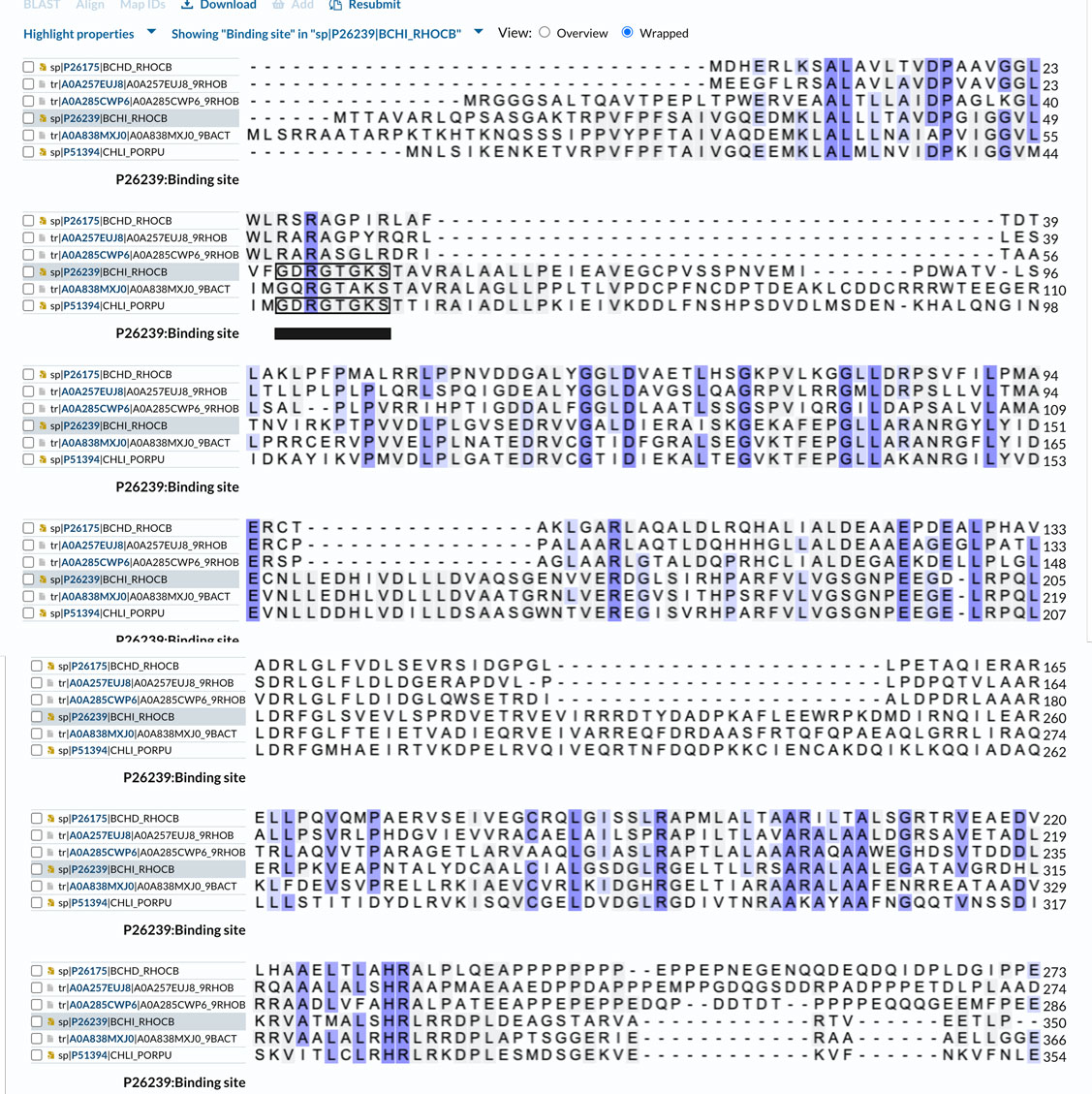
For the comparison, I have made an alignment that included BchI sequences. The sequence similarity is about 29%, which is not very high. The alignment (click to see it, only part of the BchD sequence is shown) also shows that the ATP binding site residues (the P-loop motif, marked on the alignment) of BchI are not conserved in BchD. This means that BchD does not hydrolyze ATP. However, it is still possible that ATP may bind to the protein and trigger oligomerization of the complex (it does for BchI). The alignment also shows large insertions in the BchI sequence compared to BchD. This is another reason for submitting the predicted structure of BchD to a fold recognition server. This way, we can verify that we indeed have a similar fold.
Final notes
This tutorial demonstrates what can be done using the sequence analysis tools at UniProt and other servers, and including a predicted AlphaFold structure. One of the problems of AlphaFold predictions is that the structures are not classified in the CATH database. Hopefully, at some moment, this will change. Until then, we need to rely on our own analysis for the identification of domain fold in the predicted structures.
I am sure that there are many other ways to combine sequence and structure analysis. In addition, our analysis depends on our aim and what we try to achieve. I hope that this introduction will help you in your search for information.
This tutorial demonstrates what can be done using the sequence analysis tools at UniProt and other servers, and including a predicted AlphaFold structure. One of the problems of AlphaFold predictions is that the structures are not classified in the CATH database. Hopefully, at some moment, this will change. Until then, we need to rely on our own analysis for the identification of domain fold in the predicted structures.
I am sure that there are many other ways to combine sequence and structure analysis. In addition, our analysis depends on our aim and what we try to achieve. I hope that this introduction will help you in your search for information.
The FASTA format
As noted above, many applications require the amino acid sequence to be in FASTA format. The FASTA format includes the amino acid sequence in one-letter code, usually with 60 letters/line. Most important is the sign ">," “larger than,” on the first line. Alignment programs at the EBI MSA server will use the text after the >-sign on that line as the alignment title for the sequence. For convenience, one could write the name of the protein on that row, which would be helpful as a sequence identifier after running the alignment. BchI sequence in FASTA format is shown below:
>sp|P26239|BCHI_RHOCB Magnesium-chelatase 38 kDa subunit OS=Rhodobacter capsulatus (strain ATCC BAA-309 / NBRC 16581 / SB1003) OX=272942 GN=bchI PE=1 SV=1
MTTAVARLQPSASGAKTRPVFPFSAIVGQEDMKLALLLTAVDPGIGGVLVFGDRGTGKST
AVRALAALLPEIEAVEGCPVSSPNVEMIPDWATVLSTNVIRKPTPVVDLPLGVSEDRVVG
ALDIERAISKGEKAFEPGLLARANRGYLYIDECNLLEDHIVDLLLDVAQSGENVVERDGL
SIRHPARFVLVGSGNPEEGDLRPQLLDRFGLSVEVLSPRDVETRVEVIRRRDTYDADPKA
FLEEWRPKDMDIRNQILEARERLPKVEAPNTALYDCAALCIALGSDGLRGELTLLRSARA
LAALEGATAVGRDHLKRVATMALSHRLRRDPLDEAGSTARVARTVEETLP
As noted above, many applications require the amino acid sequence to be in FASTA format. The FASTA format includes the amino acid sequence in one-letter code, usually with 60 letters/line. Most important is the sign ">," “larger than,” on the first line. Alignment programs at the EBI MSA server will use the text after the >-sign on that line as the alignment title for the sequence. For convenience, one could write the name of the protein on that row, which would be helpful as a sequence identifier after running the alignment. BchI sequence in FASTA format is shown below:
>sp|P26239|BCHI_RHOCB Magnesium-chelatase 38 kDa subunit OS=Rhodobacter capsulatus (strain ATCC BAA-309 / NBRC 16581 / SB1003) OX=272942 GN=bchI PE=1 SV=1
MTTAVARLQPSASGAKTRPVFPFSAIVGQEDMKLALLLTAVDPGIGGVLVFGDRGTGKST
AVRALAALLPEIEAVEGCPVSSPNVEMIPDWATVLSTNVIRKPTPVVDLPLGVSEDRVVG
ALDIERAISKGEKAFEPGLLARANRGYLYIDECNLLEDHIVDLLLDVAQSGENVVERDGL
SIRHPARFVLVGSGNPEEGDLRPQLLDRFGLSVEVLSPRDVETRVEVIRRRDTYDADPKA
FLEEWRPKDMDIRNQILEARERLPKVEAPNTALYDCAALCIALGSDGLRGELTLLRSARA
LAALEGATAVGRDHLKRVATMALSHRLRRDPLDEAGSTARVARTVEETLP