Protein Folds, Domains & Classification. The CATH Database
In the previous section, I discussed the definition of a domain and some examples of domains and folds. Here we continue the discussion about fold classification. Detailed analysis of the fold of a protein can provide deeper insights into its function and evolutionary history, which often may be difficult to achieve based on amino acid sequence analysis alone. A study of the relationships between the amino acid sequence and the fold may also provide insights into protein structure and function principles. In addition, it may aid in the design of new proteins with predefined structure and activity.
The first step in classification is defining the secondary structure of the protein or protein domains. This is routinely done when a new structure is deposited with the Protein Data Bank (PDB), but also by graphics programs that help us visualize protein structures. All PDB entries contain a detailed description of the secondary structure of the protein, including the name of the first and last amino acid residues of each helix and β-strand. The second requirement of classification is the definition of the domains in a protein. A domain is the primary "unit" of classification. As mentioned earlier, the same domain type may be found in many unrelated proteins, for example, the nucleotide-binding Rossmann fold domain. For this reason, it is not meant to base the classification on the whole protein if it contains more than one domain.
The PDB coordinate file does not provide information on the domain content of a protein, but it usually provides links to databases where this information can be found. The primary database on domains and domain classification is CATH (C-class, A-Architecture, T-Topology, H-Homologous superfamily). CATH gives information on the domain content of each protein and a detailed description of each domain. The assignment procedure includes:
•Assignment of a Class to each domain (essentially refers to the secondary structure content - alpha, beta, or alpha/beta proteins)
•Assignment of Architecture (the arrangement of secondary structure elements in space, irrespective of connectivity between them). The amino acid sequences within a particular architecture class are not necessarily homologous - common evolutionary origin is not required.
•Assignment of Topology. Topology is what I was referring to fold. Here connectivity between secondary structure elements is considered. Proteins with a similar fold do not need to have a common evolutionary origin.
•Assignment of Homologous superfamily. A homologous superfamily defines a group of proteins that appear to be homologous (have common evolutionary origin), even without significant sequence similarity.
We will look at some examples of CATH classification to explain these definitions.
More about domains - multidomain proteins
As noted earlier, some proteins contain a single domain, while others may have several domains. Below are examples of a one-domain (hemoglobin 1IT2, on the left), and a 3-domain protein (pyruvate kinase 1PKN, on the right):
The first step in classification is defining the secondary structure of the protein or protein domains. This is routinely done when a new structure is deposited with the Protein Data Bank (PDB), but also by graphics programs that help us visualize protein structures. All PDB entries contain a detailed description of the secondary structure of the protein, including the name of the first and last amino acid residues of each helix and β-strand. The second requirement of classification is the definition of the domains in a protein. A domain is the primary "unit" of classification. As mentioned earlier, the same domain type may be found in many unrelated proteins, for example, the nucleotide-binding Rossmann fold domain. For this reason, it is not meant to base the classification on the whole protein if it contains more than one domain.
The PDB coordinate file does not provide information on the domain content of a protein, but it usually provides links to databases where this information can be found. The primary database on domains and domain classification is CATH (C-class, A-Architecture, T-Topology, H-Homologous superfamily). CATH gives information on the domain content of each protein and a detailed description of each domain. The assignment procedure includes:
•Assignment of a Class to each domain (essentially refers to the secondary structure content - alpha, beta, or alpha/beta proteins)
•Assignment of Architecture (the arrangement of secondary structure elements in space, irrespective of connectivity between them). The amino acid sequences within a particular architecture class are not necessarily homologous - common evolutionary origin is not required.
•Assignment of Topology. Topology is what I was referring to fold. Here connectivity between secondary structure elements is considered. Proteins with a similar fold do not need to have a common evolutionary origin.
•Assignment of Homologous superfamily. A homologous superfamily defines a group of proteins that appear to be homologous (have common evolutionary origin), even without significant sequence similarity.
We will look at some examples of CATH classification to explain these definitions.
More about domains - multidomain proteins
As noted earlier, some proteins contain a single domain, while others may have several domains. Below are examples of a one-domain (hemoglobin 1IT2, on the left), and a 3-domain protein (pyruvate kinase 1PKN, on the right):
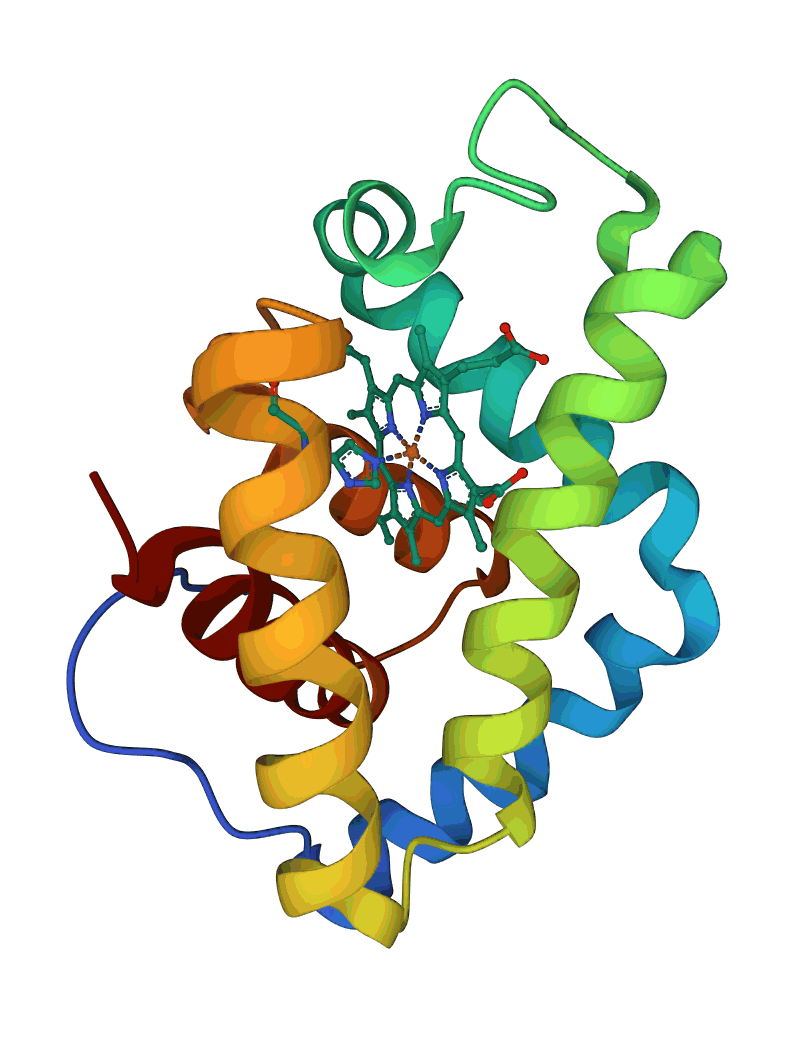
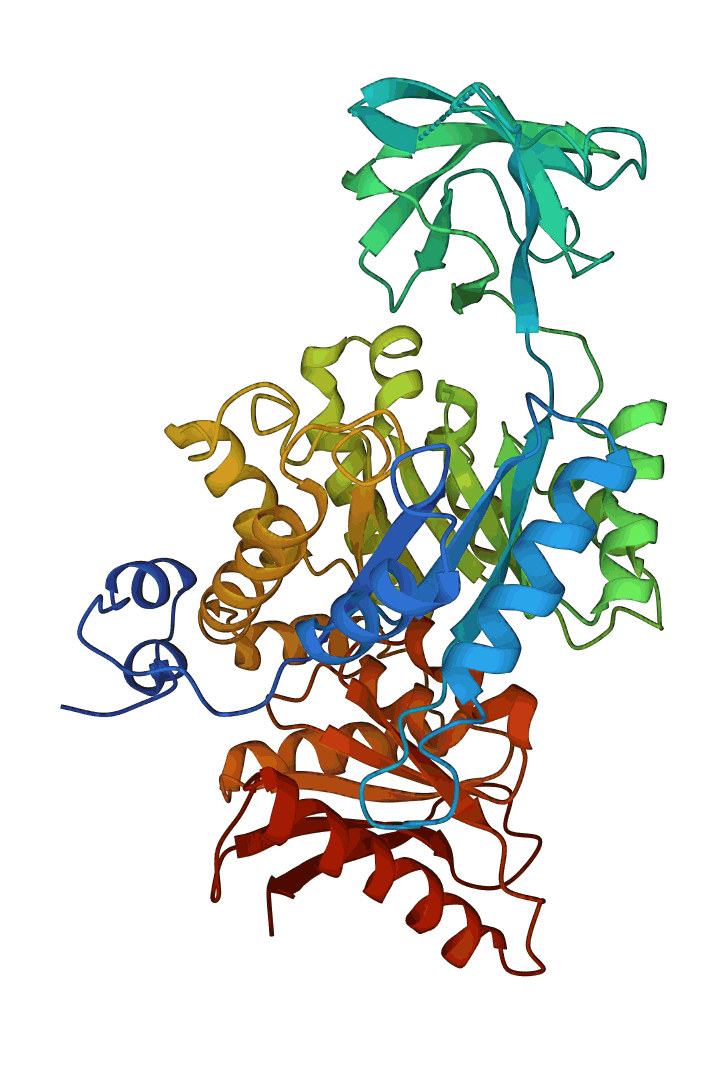
The three domains in pyruvate kinase are well separated and have different folds. For example, the top domain in the Figure above is a β-sheet domain, while the other two are of alpha/beta type (see the respective Proteopedia page for details).
In most organisms, these two proteins' functional unit is tetrameric (containing four subunits). In the case of hemoglobin, there will be four molecules (and four domains) in each active unit, while the functional unit of pyruvate kinase will contain 12 domains.
A multidomain protein - CATH classification of pyruvate kinase
Performing a search with PDB ID 1T5A (the tetrameric human pyruvate kinase) in CATH will return the following result for the three domains:
In most organisms, these two proteins' functional unit is tetrameric (containing four subunits). In the case of hemoglobin, there will be four molecules (and four domains) in each active unit, while the functional unit of pyruvate kinase will contain 12 domains.
A multidomain protein - CATH classification of pyruvate kinase
Performing a search with PDB ID 1T5A (the tetrameric human pyruvate kinase) in CATH will return the following result for the three domains:

1t5aA01 corresponds to chain A, domain 01 (there are four chains in PDB entry 1t5a, each containing three domains). The numbers associating each protein domain with a CATH superfamily are 3.40.1380.20, 3.20.20.60, and 2.40.33.10.
Each superfamily and each domain have its page in CATH. If we, for example, click on 3.40.1380.20, we get to the information page for the domain (shown below) with the classification assignment:
Class: 3, Alpha Βeta,
Architecture: 3.40, 2-Layer(aba) Sandwich,
Topology: 3.40.1380, Pyruvate Kinase; Chain A, domain 1.
Homologous superfamily: 3.40.1380.20, Pyruvate Kinase C-terminal domain.
Each superfamily and each domain have its page in CATH. If we, for example, click on 3.40.1380.20, we get to the information page for the domain (shown below) with the classification assignment:
Class: 3, Alpha Βeta,
Architecture: 3.40, 2-Layer(aba) Sandwich,
Topology: 3.40.1380, Pyruvate Kinase; Chain A, domain 1.
Homologous superfamily: 3.40.1380.20, Pyruvate Kinase C-terminal domain.
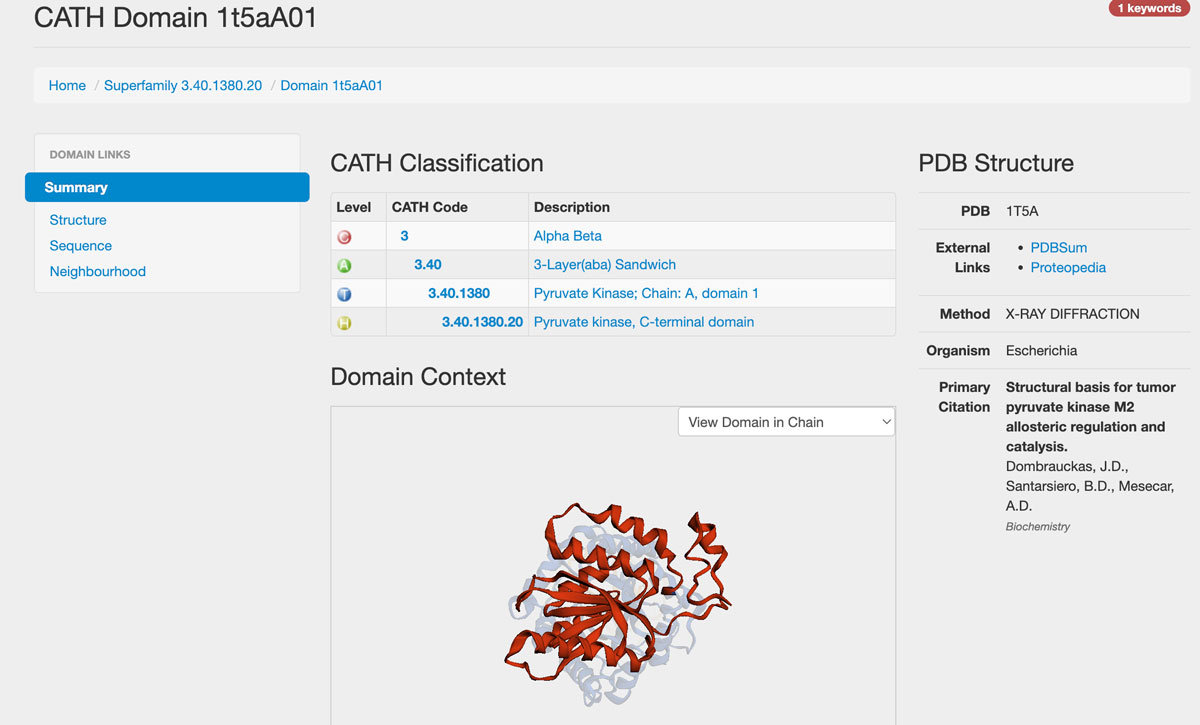
From here, we can continue exploring the domain's details and the superfamily it belongs to. For example, suppose we click "Pyruvate Kinase C-terminal", which is the name of the superfamily, or click the CATH code 3.40.1380.20. In that case, we will be taken to the page of this Homologues superfamily (can be accessed here):
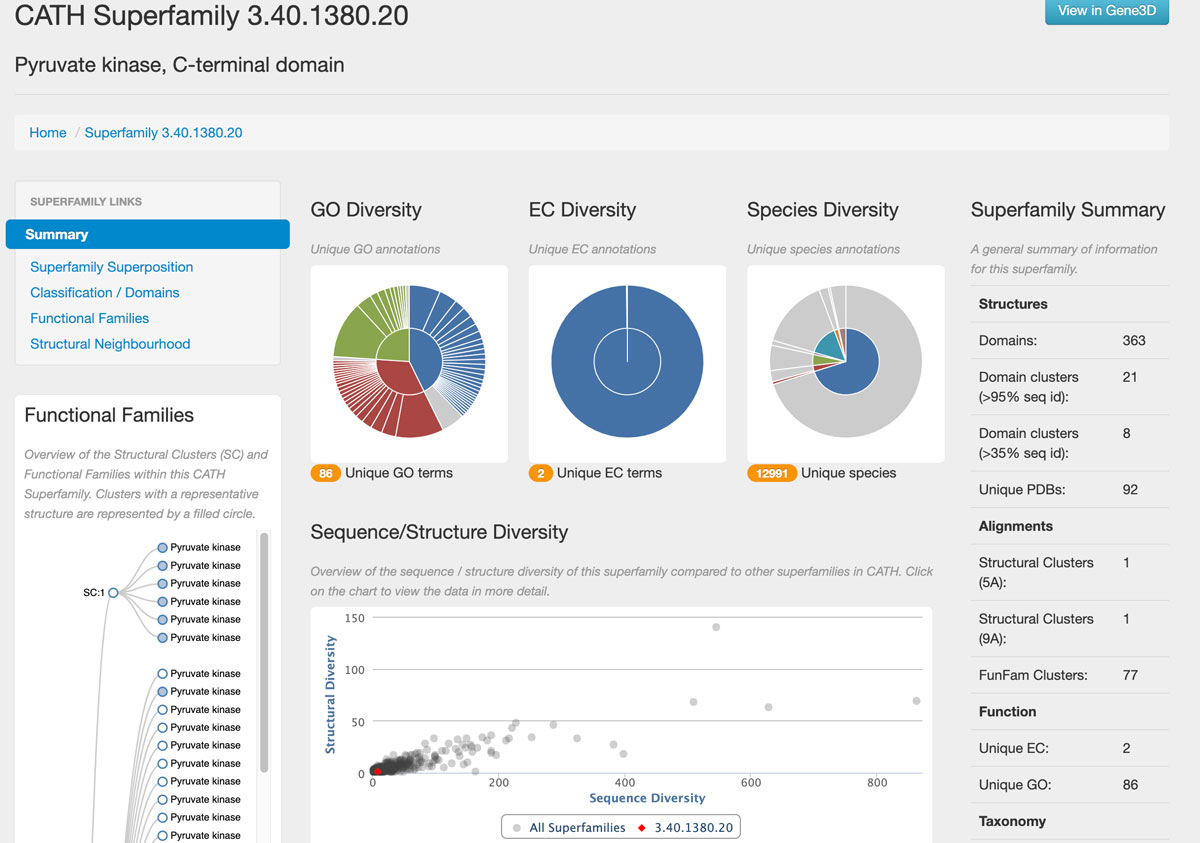
On the right is a summary of some data related to this superfamily. We can see that there is only one structural cluster, meaning this superfamily has low structural diversity. This can also be seen in the graph. It compares the Sequence/Structure diversity of the pyruvate kinase C-terminal domain superfamily with all other superfamilies in CATH. Here the red spot corresponds to 3.40.1380.20. Note that clicking at any spot will show the name of the corresponding superfamily, the numbers of sequence families, and the structural clusters within the superfamily. This low diversity is also apparent from the EC (Enzyme Commission) diversity chart - only two EC numbers are associated with this domain.
Using the CATH superfamily code, we can also search the Mechanism and Catalytic Site Atlas that will give us a summary of the enzymatic reaction and some links to other sites with details of pyruvate kinase function, like Interpro.
The "superfamily superposition" link on the left gives an image that shows how well the structures of this domain are conserved within the superfamily (image below).
Using the CATH superfamily code, we can also search the Mechanism and Catalytic Site Atlas that will give us a summary of the enzymatic reaction and some links to other sites with details of pyruvate kinase function, like Interpro.
The "superfamily superposition" link on the left gives an image that shows how well the structures of this domain are conserved within the superfamily (image below).
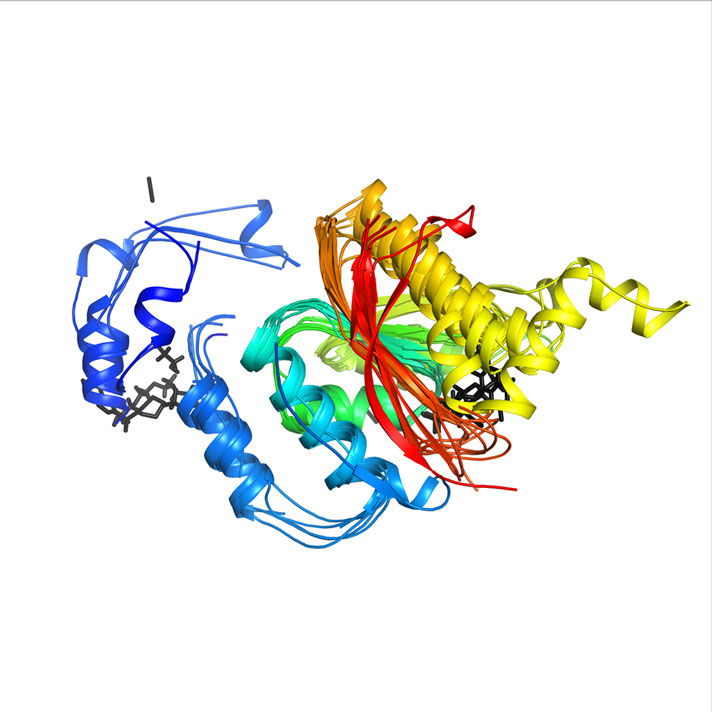
Other options to explore here include functional families, structural neighborhoods, etc. We will use some of these later. For now, you are welcome to do your research and compare the findings for this domain with the same features of the other two domains. You will discover, for example, that the second domain, 3.20.20.60 belongs to the phosphoenolpyruvate-binding domain superfamily that has considerably higher diversity than the C-terminal domain.
There are many questions one can ask while exploring this database. For example, it would be interesting to know if the structures of these three domains are equally conserved within their families, what functions (enzymatic activity or something else) are assigned to them, and to compare the conservation of sequence and structure. This will give exciting insights into the structure and function of pyruvate kinase.
There are also two links on the right side of the page, one to the Proteopedia page that describes pyruvate kinase and the second to PDBsum. In the next section, I will show some valuable features of PDBsum.
There are many questions one can ask while exploring this database. For example, it would be interesting to know if the structures of these three domains are equally conserved within their families, what functions (enzymatic activity or something else) are assigned to them, and to compare the conservation of sequence and structure. This will give exciting insights into the structure and function of pyruvate kinase.
There are also two links on the right side of the page, one to the Proteopedia page that describes pyruvate kinase and the second to PDBsum. In the next section, I will show some valuable features of PDBsum.