Protein Structure Databases: The Protein Databank PDB, PDBe & PDBsum
There are many structural bioinformatics-related resources on the Internet. Some are general, while others are dedicated to specific aspects of proteins and protein families, particular functions, metabolic pathways, etc. Proteopedia is an excellent example of a public educational database, to which you may find several links on this site. It provides in-depth discussions of the structural and functional aspects of many proteins. Here we focus on the databases that are often used and are essential for the fields of structural biology and structural bioinformatics - the RSCB Protein Databank (PDB) and two European versions, PDBe and PDBsum.
The first questions to ask when exploring a protein should probably be - is there a 3D structure, and where to get the coordinate file? For an extended period, the primary database for protein structures was the RSCB Protein Data Bank, created at the beginning of the 1970-ties. Only a few structures existed then; the only experimental method for protein structure determination was protein X-ray crystallography. As we can see from the image below, starting from the 1990ties, PDB content growth has been awe-inspiring:
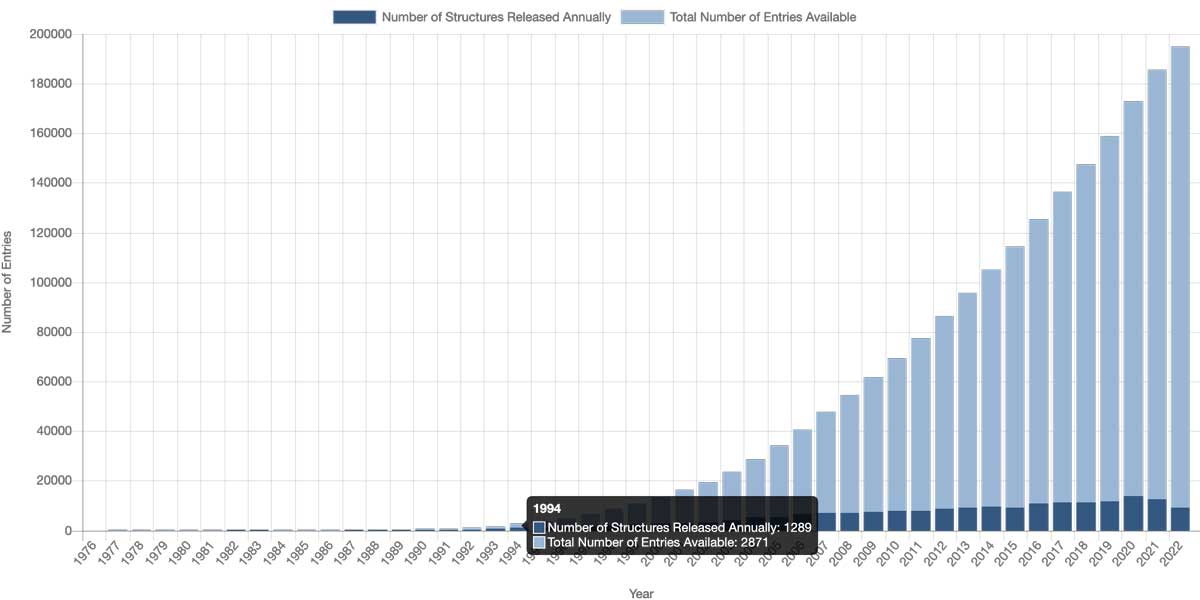
The structural revolution
There are, of course, several reasons for the structural revolution. One of them was that cloning techniques started to enter the lab, and the number of different proteins and their quantity available for crystallization increased drastically. Before the cloning era, proteins were purified directly from cells, which substantially limited availability − there is always a limited number of copies of a particular protein in a cell. Therefore, obtaining a few milligrams of protein for crystallization required large cell volumes. Cloning solved the problem; proteins could be expressed in large quantities and purified for crystallization. Another essential factor was the introduction of synchrotron radiation for X-ray data collection. Several synchrotrons worldwide currently provide high intensity X-rays for quality X-ray data collection. In addition, synchrotrons reduced the time required for the optimization of crystallization conditions.
In the early days of crystallography, we needed to optimize the crystallization conditions to grow crystals large enough for the relatively low-intensity laboratory X-ray sources. The third factor, I believe, was the introduction of low-cost personal computers with ever-increasing computational and graphics processing power. Cheaper computers also meant new software, which became much more user-friendly. In the middle of the 1980-ties, a proper graphics monitor with a computer, which was needed for model building and refinement, would cost around 100 k dollars, obviously unaffordable for personal use for people interested in science. Now a Windows PC or a Mac is all we need. Then came the era of structural genomics - large consortia were formed to develop new technologies for crystallization and solving large numbers of protein structures. With the increasing number of solved structures, the number of protein databases increased, and new tools for analyzing protein sequences and structures were rapidly developed.
Although the number of structures in the PDB is rapidly increasing, one should remember that far from all PDB entries are unique. There are many entries of the same protein in the database - some are mutant variants, others may be complexes with ligands (substrate analogs, inhibitors, cofactors), complexes with other proteins, etc. This may be a source of confusion when we try to fetch a structure from the PDB - which one to choose if there are many entries of the same protein? For our purposes, we also need to remember that not all structures in the PDB are created equal, and we need to identify the one with the best available quality (see discussion of structure quality).
Using the PDB, we can easily find the structure of the protein of interest and assess its quality. We need to type the name of the protein into the search window on the PDB site. Generally, one gets many hits, some of which would be unrelated to the search. PDBsum and PDBe (PDB Europe) usually give more narrowed search results. It is also possible to refine the search using the options provided at the PDB site.
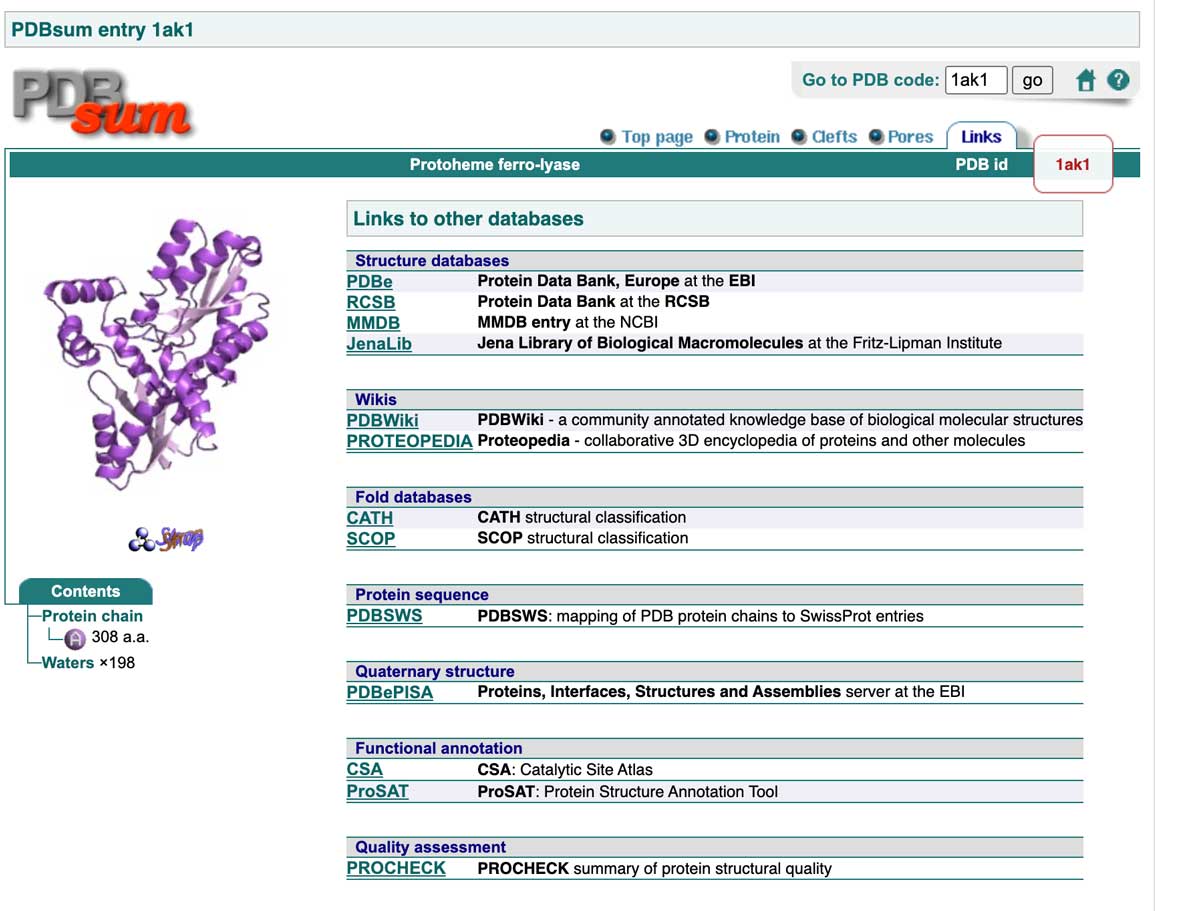
PDB, PDBe, and PDBsum provide plenty of additional data, including links to other databases where more information can be found. Below is an example from the PDBsum link page (for mobile view, please click here).
In the early days of crystallography, we needed to optimize the crystallization conditions to grow crystals large enough for the relatively low-intensity laboratory X-ray sources. The third factor, I believe, was the introduction of low-cost personal computers with ever-increasing computational and graphics processing power. Cheaper computers also meant new software, which became much more user-friendly. In the middle of the 1980-ties, a proper graphics monitor with a computer, which was needed for model building and refinement, would cost around 100 k dollars, obviously unaffordable for personal use for people interested in science. Now a Windows PC or a Mac is all we need. Then came the era of structural genomics - large consortia were formed to develop new technologies for crystallization and solving large numbers of protein structures. With the increasing number of solved structures, the number of protein databases increased, and new tools for analyzing protein sequences and structures were rapidly developed.
Although the number of structures in the PDB is rapidly increasing, one should remember that far from all PDB entries are unique. There are many entries of the same protein in the database - some are mutant variants, others may be complexes with ligands (substrate analogs, inhibitors, cofactors), complexes with other proteins, etc. This may be a source of confusion when we try to fetch a structure from the PDB - which one to choose if there are many entries of the same protein? For our purposes, we also need to remember that not all structures in the PDB are created equal, and we need to identify the one with the best available quality (see discussion of structure quality).
Using the PDB, we can easily find the structure of the protein of interest and assess its quality. We need to type the name of the protein into the search window on the PDB site. Generally, one gets many hits, some of which would be unrelated to the search. PDBsum and PDBe (PDB Europe) usually give more narrowed search results. It is also possible to refine the search using the options provided at the PDB site.
PDB, PDBe, and PDBsum provide plenty of additional data, including links to other databases where more information can be found. Below is an example from the PDBsum link page (for mobile view, please click here).
The asymmetric unit - what is it?
We also need to remember that PDB files contain the so-called asymmetric unit of the crystal. The functional biological unit (the quaternary structure) in solution may contain several subunits of the same protein, arranged as dimers, trimers, or larger-order oligomers, as discussed earlier. Often the subunits in these quaternary structures in solution are related by some symmetry - for example, two-, three- or four-fold rotation. When crystallized, the oligomer symmetry axis may become a crystallographic symmetry axis. This means that a monomer within a dimer, a trimer, or a tetramer, becomes an asymmetric unit of the crystal.
Why is it an asymmetric unit? When the molecules are crystallized, they are arranged in the space lattices of the crystal. Within this lattice, all molecules are ordered and related to each other by crystallographic symmetry operations of the symmetry group of that crystal (the possible symmetry groups are listed in a book called International Tables for Crystallography). A symmetry operation represents a mathematical transformation that, when applied to the coordinates of one molecule, will transform it to its symmetry-related mate in the crystal lattice. This is what we mean when we say that molecules in the crystal lattice are arranged according to certain symmetry operations. By applying symmetry transformation to the asymmetric unit, we can generate all other molecules in the crystal. For example, a trimer may be easily generated by applying the mathematical operations of the 3-fold rotation axis. For this reason, calculations in crystallography are performed using only the asymmetric unit of the crystal. Ideally, all other molecules in the crystal will be precisely similar.
This is reflected in the content of PDB files, which only contain the atomic coordinates of the asymmetric unit. However, the PDB server reconstructs the biological unit when it is known to be different from the asymmetric unit. If we need the biological unit, we may choose it when viewing the 3D structure in the graphics display or when downloading the coordinate file.
For clarity, the concept of the asymmetric unit is illustrated in the image below:
Why is it an asymmetric unit? When the molecules are crystallized, they are arranged in the space lattices of the crystal. Within this lattice, all molecules are ordered and related to each other by crystallographic symmetry operations of the symmetry group of that crystal (the possible symmetry groups are listed in a book called International Tables for Crystallography). A symmetry operation represents a mathematical transformation that, when applied to the coordinates of one molecule, will transform it to its symmetry-related mate in the crystal lattice. This is what we mean when we say that molecules in the crystal lattice are arranged according to certain symmetry operations. By applying symmetry transformation to the asymmetric unit, we can generate all other molecules in the crystal. For example, a trimer may be easily generated by applying the mathematical operations of the 3-fold rotation axis. For this reason, calculations in crystallography are performed using only the asymmetric unit of the crystal. Ideally, all other molecules in the crystal will be precisely similar.
This is reflected in the content of PDB files, which only contain the atomic coordinates of the asymmetric unit. However, the PDB server reconstructs the biological unit when it is known to be different from the asymmetric unit. If we need the biological unit, we may choose it when viewing the 3D structure in the graphics display or when downloading the coordinate file.
For clarity, the concept of the asymmetric unit is illustrated in the image below:
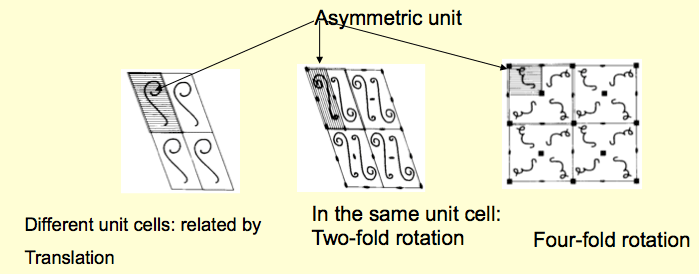
In the left Figure, the asymmetric unit of a crystal that is just "one subunit" is shown. All molecules in the lattice are related to each other by simple translation. In the example, in the middle, there are two subunits in the unit cell related to each other by a two-fold rotation axis (180 degrees of rotation around the axis). This suggests that the protein in the solution (the biological unit) could be a dimer. The third example on the right shows that a 4-fold crystallographic symmetry relates the molecules in the unit cell. Again, this suggests that the biological unit in solution is a tetramer. In all these examples, the asymmetric unit is a monomer, but we also may have a dimer, trimer, etc., in the asymmetric unit. All the symmetry operations described in this case will be applied to the whole dimer, trimer, etc. Of course, we need additional experiments to verify the protein's oligomeric status in the solution.