Introduction To Structure-Based Drug Design
In this chapter, we outline the small-molecule drug design process, in which the protein’s three-dimensional structure is extensively used at all initial stages of the project.
The Drug Market
To illustrate the relationship between small-molecule drugs and biologics, we can review the findings of a survey conducted in 2006 on marketed drugs. This survey revealed that out of 1,357 unique drugs, 1,204 were small-molecule drugs, while 166 were biological agents, primarily antibodies. The total number of drug targets (derived from human and pathogenic organisms) was 324 (Overington, Al-Lazikani & Hopkins, 2006). A more recent study by the same research group, published in 2016, found a significant increase in the number of drug targets, totaling 893, with 667 derived from human sources. The total number of FDA-approved drugs has risen to 1,578, including 195 biologics (Santos et al., 2016). The analysis indicates that while small-molecule drugs take the largest market share, the absolute number of biologics is growing, even if their relative proportion remains smaller. It’s important to note that various methods can be used to conduct these studies, and different researchers may arrive at different figures. However, since the studies cited here were conducted by the same group of authors, the reliability of the comparison is enhanced.
An Outline of Small-Molecule Drug Discovery & Design
Before initializing a drug discovery project, we must study the disease mechanism and identify an appropriate target for treatment. Most small-molecule drugs are designed to target proteins. Although drugs can also target nucleic acids, their application in disease treatment has been limited due to various factors, such as toxicity and the difficulty of achieving high specificity. Here, we will focus on small-molecule drugs, although there is a category of so-called biological drugs also known as biologics. One prominent example is insulin, but most are monoclonal antibodies. For a discussion on the role of structural biology in antibody drug design, please take a look at a series of posts on my company blog.
The question we want to ask now is: How can structural biology and structural bioinformatics help us in drug discovery? The schematic image below, showing the different phases of the drug discovery process, indicates that structural biology and bioinformatics tools may be helpful in the first three phases of the process: initial target identification and validation, hit/lead generation, and lead optimization. Below we discuss each phase in more detail.
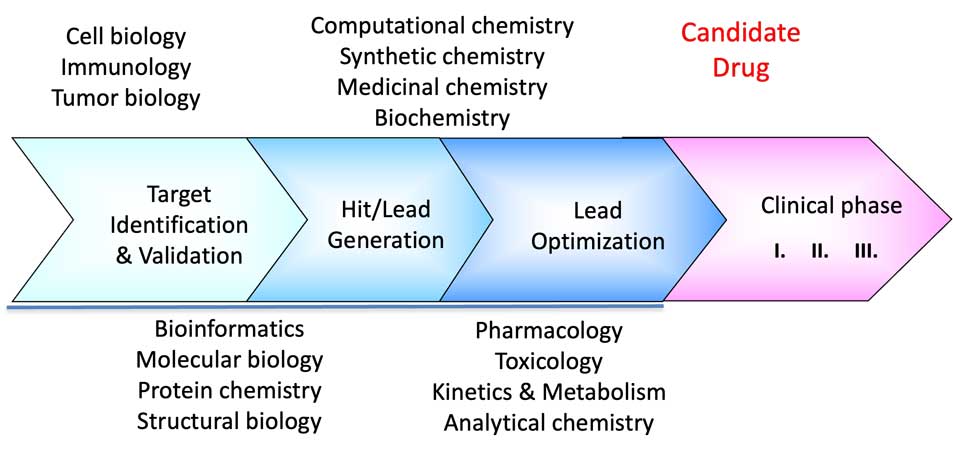
1- Target Identification & Validation
As its name suggests, this phase aims to identify the appropriate drug target involved in a disease. Typically, a target is a specific molecule in the human body (or from a pathogen, such as bacteria). It can be an enzyme in a biochemical pathway, a protein within a cellular signaling pathway, a receptor, an ion channel, or similar. For instance, headache tablets like aspirin target the enzyme cyclooxygenase (see structure-based drug discovery example later in this chapter). Statins, which reduce cholesterol levels in the body, target HMG-CoA reductase, an enzyme involved in cholesterol synthesis, while opioids target opioid receptors in the brain. Additionally, it could be a structural protein, such as those that make up bacterial cell walls, which are targeted by antibiotics. Target identification and verification necessitate thorough studies of the molecular biology and biochemistry of the disease.
At this phase, three-dimensional structure and structural bioinformatics can assist in the detailed study and analysis of the protein in question. This may involve assessing potential target draggability by identifying functional regions such as the active site, co-factor binding, allosteric sites, or surfaces involved in protein-protein interactions (PPI). Analyzing the relationships between sequence and structure can help elucidate the effects of mutations on the protein’s activity and examine its evolutionary history.
2- Hit Identification & Lead Generation
The hit identification and lead generation phase seeks to obtain a compound or a series of compounds ready to enter the lead optimization and drug development phase, respectively. During the hit identification stage, we identify molecules that can bind to the target and produce a biological effect. Central to hit identification are various high-throughput compound library screening techniques that utilize robotics, biophysical, biochemical, or cell-based assays to validate the activity of the compounds. For structure-based drug design, hit compounds are crystallized in complex with the protein target, providing a detailed view of the molecular interactions within the ligand binding site of the protein. This, in turn, facilitates further optimization of the compounds toward lead molecules and candidate drugs. The hit-to-lead process, also referred to as lead generation, focuses on enhancing various properties of the molecule, including solubility, affinity to the target, and ADME (Absorption, Distribution in the body, Metabolism, and Excretion) profile, to generate a candidate drug, a drug-like molecule. This process is usually carried out in close collaboration between proficient medicinal chemistry and structural biology teams. The significance of the availability of three-dimensional structural data in all these processes cannot be overestimated. In the following sections, we provide more details on hit identification and compound library screening, as well as fragment-based drug design (FBDD).
Computational methods with enhanced AI capabilities are essential in modern drug discovery and design. We can conduct screenings using virtual libraries that contain millions of compounds. The advantage is that the compounds can be synthesized or purchased only after demonstrating some binding efficiency in computer screenings. ADME prediction tools can also help prioritize compounds with favorable ADME profiles. AI can also assist in predicting analogs to refine structure-activity relationships (SAR). At this stage, in vitro toxicity may also be assessed. This may include cytotoxicity, genotoxicity, off-target effects (interactions with other proteins), and toxicity in mice.
3- Lead Optimization To Candidate Drug
Using the lead series obtained in the previous phase, we engage in iterative cycles of computational modeling, chemical modification, and biological testing to identify a so-called candidate drug (CD), an optimized lead molecule that can enter phase I clinical trials. A candidate drug should possess improved parameters and meet the following criteria:
- Potency – low nm to μM (the lower the better), activity against target
- Selectivity – minimal off-target effects (due to binding to other proteins or other structures in cells)
- ADMET profile – optimal pharmacokinetics and low toxicity in preclinical studies
- Efficacy – demonstrating activity in the disease model (usually animals)
- Synthetic feasibility – cost-effective synthesis demonstrated in the laboratory
- Intellectual property value – patentable chemical scaffold
Finally, we should note that in a case when known inhibitors of the enzyme already exist (an excellent place to look for those can be the PDBe Chemical Components Library), the structures of the inhibitors in complex with the protein can be used for mapping the interactions within the binding site and for building a so-called pharmacophore model for the binding site. Such models can be used, among other things, in filtering compound libraries before screening for potential binders. There is no use of compounds that would not fit into the protein binding site, for example, due to bad shape complementarity, absence of groups that would interact with the binding-site amino acids, presence of charged groups in cases of a hydrophobic ligand site, etc.