Compound Library Screening, Hit Identification & Optimization
Identifying an organic molecule which at some moment, after clinical trials, will provide efficient and safe disease treatment is not an easy task. The space of small organic molecules is enormous and contains an endless number of compounds. To limit the number of molecules in the libraries used for screening, we can apply filtering, which was briefly mentioned in the introduction.
Library filtering by analyzing protein-ligand interactions
At an initial filtering step, the most prominent factors to consider are shape and physical-chemical complementarity between the ligand and the binding site. This involves the molecular interactions that contribute to ligand binding stabilization:
• Hydrophobic interactions
• Hydrogen bonds
• Electrostatic forces (salt bridges)
• Helix dipoles
• Aromatic-aromatic interactions
• Entropic effects
• Water molecules
The relative importance of these interactions depends on the energy involved. For example, hydrogen bond energy is in the range of 2-10 kcal/mol., while electrostatic interaction energy is in the range of 3-5 kcal/mol, depending on the distance and polarity of the environment. By analyzing the interactions the protein binding site may provide, we can choose library compounds that may satisfy these interactions. This will substantially reduce the total number of potential binders. The set of potential interactions within the ligand-binding site helps in the construction of a pharmacophore model that can be used, e.g., in the virtual screening of a compound library.
Library filtering by analyzing protein-ligand interactions
At an initial filtering step, the most prominent factors to consider are shape and physical-chemical complementarity between the ligand and the binding site. This involves the molecular interactions that contribute to ligand binding stabilization:
• Hydrophobic interactions
• Hydrogen bonds
• Electrostatic forces (salt bridges)
• Helix dipoles
• Aromatic-aromatic interactions
• Entropic effects
• Water molecules
The relative importance of these interactions depends on the energy involved. For example, hydrogen bond energy is in the range of 2-10 kcal/mol., while electrostatic interaction energy is in the range of 3-5 kcal/mol, depending on the distance and polarity of the environment. By analyzing the interactions the protein binding site may provide, we can choose library compounds that may satisfy these interactions. This will substantially reduce the total number of potential binders. The set of potential interactions within the ligand-binding site helps in the construction of a pharmacophore model that can be used, e.g., in the virtual screening of a compound library.
Structure-based design strategies
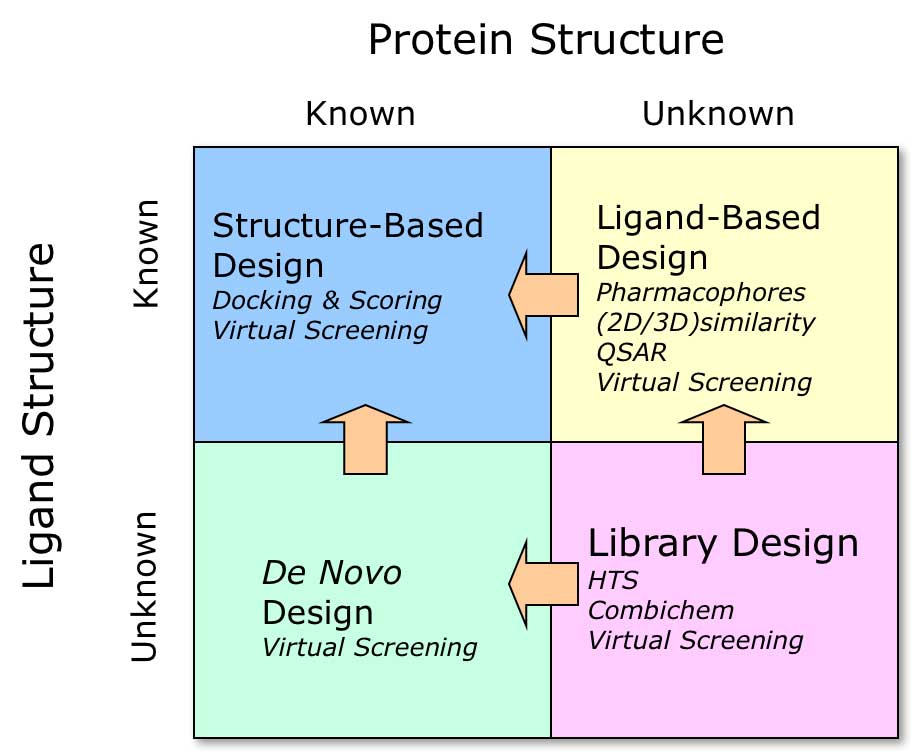
Depending on availability of information, different drug discovery strategies may be followed. Detailed discussion of the strategies in the image above can be found in our blog post Structure-Based Discovery Strategies
Library filtering - Lipinski's rule of 5
Apart from the above factors, some empirical rules have been developed for limiting the chemical space while preparing suitable screening libraries. Lipinski and his colleagues have defined the so-called Lipinski's rule of 5. These rules state that an orally active drug should not have more than one violation of the following criteria:
Together with a pharmacophore model, these rules will achieve considerable chemical space limitation, making the screening task much easier to handle. There are, of course, other methods that are used to limit the size of a compound library. One such method described on the next page is the use of fragment libraries.
- Mw < 500 g/mol
- Hydrogen bond acceptors < 10 (N and O)
- hydrogen bond donors < 5 (OH and NH)
- logP < 5 (logP=log ([Coctanol]/[Cwater])
Together with a pharmacophore model, these rules will achieve considerable chemical space limitation, making the screening task much easier to handle. There are, of course, other methods that are used to limit the size of a compound library. One such method described on the next page is the use of fragment libraries.
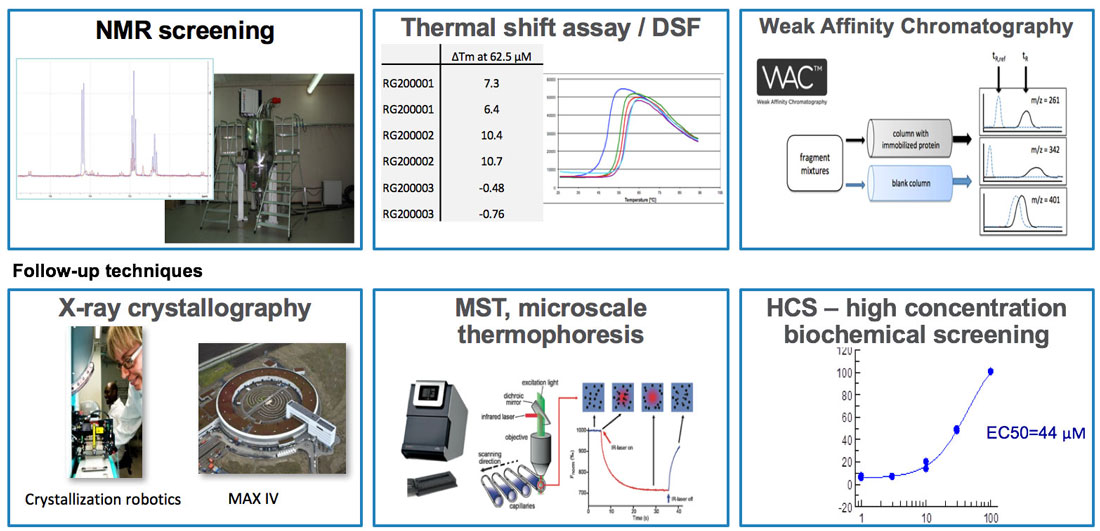
Biophysical assays: library screening using biophysical methods
Screening is used for initial hit identification after constructing a compound library. Several biophysical methods (assays) can be employed in screening compound libraries. They include:
Later, I will provide more details on X-ray crystallography, but going into the details of all other methods is not within the scope of this material. However, it is part of the subject of drug discovery, and we usually mention these methods during our course. I think it is valuable to know of their existence. I should also note that WAC screening is a proprietary method jointly owned by our company SARomics Biostructures and our partner Red Glead Discovery. You may read more about the use of WAC in fragment screening on the company homepage.
- X-ray crystallography
- NMR spectroscopy
- Surface plasmon resonance (SPR)
- Weak affinity chromatography (WAC)
- Thermal shift assay (differential scanning fluorimetry, DSF)
- Microscale thermophoresis (MST)
- Differential scanning calorimetry (DSL)
Later, I will provide more details on X-ray crystallography, but going into the details of all other methods is not within the scope of this material. However, it is part of the subject of drug discovery, and we usually mention these methods during our course. I think it is valuable to know of their existence. I should also note that WAC screening is a proprietary method jointly owned by our company SARomics Biostructures and our partner Red Glead Discovery. You may read more about the use of WAC in fragment screening on the company homepage.
Structure-based lead discovery and lead optimization
Since most initial hits usually have a low affinity towards the target, they need to be modified to improve binding energy and specificity. This is the process of lead generation and lead optimization in lead discovery. We also need a biochemical activity assay, which will allow us to assess the effect of the compounds on the protein in solution and cell cultures. Assuming that the protein's three-dimensional structure is available, the best compounds identified in screening are co-crystallized with the protein to obtain the structure of the protein-ligand complex. Analysis of the binding mode of the ligand will provide the insights required for further optimization of its structure and binding affinity. This will be the start of a structure-based drug design project. The process typically involves a repeated determination of the structures of the drug target protein in complex with many compounds. Sometimes 10th of such structures may be required before the desired molecule is made. Structural information will contribute to improving the potency and selectivity of the inhibitors. The methods of structure-activity relationships analysis (SAR) will also be helpful during this process. For those who might be interested, I provide an example of the use of the three-dimensional structure in the design of specific inhibitors of the enzyme cyclooxygenase 2 (COX2).
In cases when no experimental structure is available, predicted structures may be used to guide the initial design of new inhibitors if we, for example, use computational methods and run a virtual screening and docking of compound libraries. It should be kept in mind that predicted structures do not always provide models with accurate atomic positions. However, they may be quite useful in crystallization since they often reveal regions with long flexible loops. Such loops can be removed to produce a new protein that can be more prone to successful crystallization.
In cases when no experimental structure is available, predicted structures may be used to guide the initial design of new inhibitors if we, for example, use computational methods and run a virtual screening and docking of compound libraries. It should be kept in mind that predicted structures do not always provide models with accurate atomic positions. However, they may be quite useful in crystallization since they often reveal regions with long flexible loops. Such loops can be removed to produce a new protein that can be more prone to successful crystallization.