PDB File Format and Content
We continue the discussion of the PDB and look inside the coordinate file - it is easy to download a PDB file (and it is free!). After finding the protein of interest (1G8P in this case), we will be taken to the protein-specific page. In the right corner of that page is a drop-down menu (Download Files). When clicked, it provides several choices of different formats of the PDB file (click the image below, for mobile view, please click here). The "PDB format" file (not the gz-file, a compressed file) is a text file and can be opened by any text editor (including MS Word). Usually, the file is called a "coordinate file" because it contains a list of coordinates for all atoms of the protein structure in a conventional orthogonal coordinate system. As in any coordinate system, each atom's position is defined by its x,y,z coordinates.
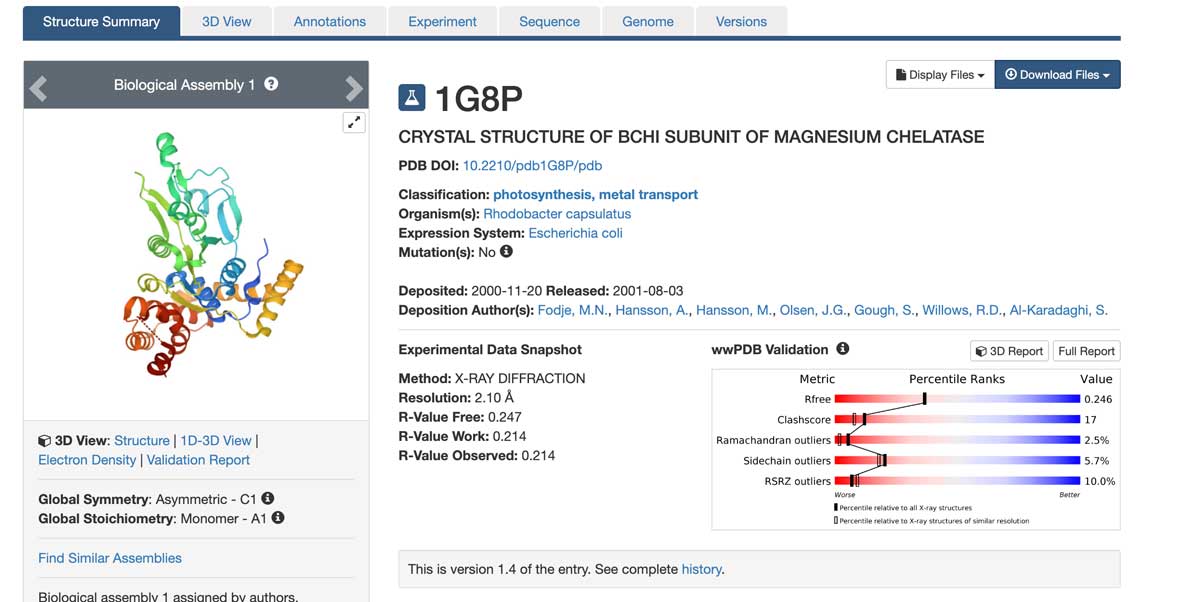
Apart from the coordinates, the file also contains essential information on the method used to solve the structure, the parameters related to the quality of the X-ray data (like resolution), as well as the symmetry operations for the specific space group of the crystal, the quality of the model geometry (deviations of bond lengths, bond angles and torsion angles from ideal values), secondary structure content, description of missing regions in the structure (a result of weak electron density due to flexibility/disorder in the structure).
The R-factor and resolution are probably the two most central parameters for the assessment of the quality of the structure. The R-factor tells us how good the fit of the structural model to the X-ray data (the electron density) is. A higher resolution of the X-ray data usually provides a better fit and results in a lower R-factor. In my experience, good quality, well-refined protein structures have a resolution of (or better than) 2.2 Å and an R-factor below 20%. At this resolution, the electron density for most atoms looks nice and well-separated from its neighbors.
Below is part of a PDB file header showing some of the data like resolution (2.1 Å), resolution range of the data (from lowest, 29.55 Å to highest, 2.10 Å), the number of reflections collected from the crystal during the X-ray experiment (22179) and the R-factor (0,214). The R-factor is ok but not great in this case. This is due to the high flexibility of parts of the structure, making reliable model building impossible. The result is that parts of the model have not been accounted for, which gives a poor fit to the experimental data.
REMARK 1
REMARK 2
REMARK 2 RESOLUTION. 2.10 ANGSTROMS.
REMARK 3
REMARK 3 REFINEMENT.
REMARK 3 PROGRAM : CNS 1.0
REMARK 3 AUTHORS : BRUNGER,ADAMS,CLORE,DELANO,GROS,GROSSE-
REMARK 3 : KUNSTLEVE,JIANG,KUSZEWSKI,NILGES, PANNU,
REMARK 3 : READ,RICE,SIMONSON,WARREN
REMARK 3
REMARK 3 REFINEMENT TARGET : ENGH & HUBER
REMARK 3
REMARK 3 DATA USED IN REFINEMENT.
REMARK 3 RESOLUTION RANGE HIGH (ANGSTROMS) : 2.10
REMARK 3 RESOLUTION RANGE LOW (ANGSTROMS) : 29.55
REMARK 3 DATA CUTOFF (SIGMA(F)) : 0.000
REMARK 3 DATA CUTOFF HIGH (ABS(F)) : 312841.620
REMARK 3 DATA CUTOFF LOW (ABS(F)) : 0.0000
REMARK 3 COMPLETENESS (WORKING+TEST) (%) : 97.9
REMARK 3 NUMBER OF REFLECTIONS : 22179
REMARK 3
REMARK 3 FIT TO DATA USED IN REFINEMENT.
REMARK 3 CROSS-VALIDATION METHOD : THROUGHOUT
REMARK 3 FREE R VALUE TEST SET SELECTION : RANDOM
REMARK 3 R VALUE (WORKING SET) : 0.214
REMARK 3 FREE R VALUE : 0.247
REMARK 3 FREE R VALUE TEST SET SIZE (%) : 10.000
REMARK 3 FREE R VALUE TEST SET COUNT : 2207
REMARK 3 ESTIMATED ERROR OF FREE R VALUE : 0.005
REMARK 3
Further down, there is a list of the secondary structure elements within the structure, also showing the first and last residue in each element:
HELIX 1 1 PRO A 22 ILE A 26 5 5
HELIX 2 2 GLN A 29 ASP A 42 1 14
HELIX 3 3 PRO A 43 GLY A 46 5 4
HELIX 4 4 ASP A 53 GLY A 57 5 5
HELIX 5 5 SER A 59 LEU A 69 1 11
HELIX 6 6 ASN A 84 ILE A 88 5 5
HELIX 7 7 SER A 114 GLY A 120 1 7
HELIX 8 8 ASP A 123 GLY A 131 1 9
HELIX 9 9 GLY A 138 ASN A 144 1 7
HELIX 10 10 GLU A 152 LEU A 156 5 5
HELIX 11 11 GLU A 157 GLY A 171 1 15
HELIX 12 12 ARG A 202 ASP A 207 1 6
HELIX 13 13 ASP A 220 ASP A 237 1 18
HELIX 14 14 ASP A 237 LEU A 263 1 27
HELIX 15 15 PRO A 264 VAL A 266 5 3
HELIX 16 16 PRO A 269 LEU A 283 1 15
HELIX 17 17 GLY A 287 GLU A 305 1 19
HELIX 18 18 GLY A 311 SER A 324 1 14
HELIX 19 19 HIS A 325 LEU A 327 5 3
HELIX 20 20 VAL A 341 LEU A 349 1 9
SHEET 1 A 5 VAL A 106 LEU A 109 0
SHEET 2 A 5 GLY A 146 ILE A 150 1 O TYR A 147 N VAL A 107
SHEET 3 A 5 PHE A 188 GLY A 194 1 O VAL A 189 N LEU A 148
SHEET 4 A 5 VAL A 48 PHE A 51 1 N VAL A 48 O LEU A 190
SHEET 5 A 5 LEU A 211 GLU A 214 1 O LEU A 211 N LEU A 49
SHEET 1 B 2 ILE A 72 VAL A 75 0
SHEET 2 B 2 VAL A 99 LYS A 102 -1 N ILE A 100 O ALA A 74
SHEET 1 C 2 ALA A 121 LEU A 122 0
SHEET 2 C 2 PHE A 135 GLU A 136 -1 N GLU A 136 O ALA A 121
SHEET 1 D 2 GLU A 172 VAL A 175 0
SHEET 2 D 2 ILE A 182 PRO A 185 -1 O ILE A 182 N VAL A 175
CRYST1 90.259 90.259 83.716 90.00 90.00 120.00 P 65 6
After the general information, the x,y,z coordinates of the atoms are listed:
ATOM 1 N ARG A 18 14.699 61.369 62.050 1.00 39.19 N
ATOM 2 CA ARG A 18 14.500 62.241 60.856 1.00 38.35 C
ATOM 3 C ARG A 18 13.762 61.516 59.729 1.00 36.05 C
ATOM 4 O ARG A 18 14.354 60.740 58.982 1.00 34.91 O
ATOM 5 CB ARG A 18 15.850 62.753 60.334 1.00 42.36 C
ATOM 6 CG ARG A 18 16.537 63.770 61.247 1.00 46.92 C
ATOM 7 CD ARG A 18 17.825 64.314 60.629 1.00 51.24 C
ATOM 8 NE ARG A 18 18.442 65.347 61.462 1.00 54.15 N
When looking at the coordinates, notice that, in this case, the structure starts at amino acid Arg 18! Amino acids from 1 to 17 are missing. The reason, as mentioned above, is poor electron density for these residues, insufficient for building them into the density (see, for example, the discussion on structure quality in homology modeling in a later chapter). Finding the correct positions for the amino acids is impossible without the guiding electron density. This shows that we need to know that many structures in the PDB may have missing parts, sometimes in loop regions. Very often, it is a side chain (or side chains) on the molecule's surface, and in the worst cases, a whole domain may be missing.
The numbers after the first record in the file, ATOM, are just sequential numbers of the atoms in the list. The atom type follows this - for example, CA means C-α, the carbon atom to which the side chain of the amino acid is attached. Next is the main chain carbon atom C, followed by the carbonyl oxygen O. Side chain atoms C-β, CG, and CD (gamma, delta, etc.), are listed according to the Greek alphabet. After the atom type, we find the name of the amino acid (ARG in this case), followed by a letter, in this file A. This letter is the so-called chain identifier. In cases when the structure consists of several polypeptide chains (e.g., as in the tetramers of hemoglobin and Pyruvate kinase discussed earlier), each chain will get its identifier, like A, B, C, etc. The three following numbers (e.g., 14.699, 61.369, 62.050 for the very first atom) are the atom's x,y,z coordinates. As mentioned above, they describe the position of each atom in an orthogonal coordinate system. If we can describe the positions of all atoms in the protein, we can draw the whole molecular structure.
In most cases, the x,y,z coordinates are followed by a number, 1. This is called occupancy. Due to local flexibility, an amino acid's side chain may have two or more different conformations. These conformations can be distinguished in the electron density map of the structure. In this case, the crystallographer will build all conformations and, for each atom, refine a parameter called occupancy (1 for full occupancy, < 1 for partial occupancies, the sum always being 1). In PDB files, these alternate conformations are marked with "ALT."
The B-factor
The numbers in the last column in the file are called the temperature factors or B-factor. The B-factor describes the displacement of the atomic positions from an average (mean) value (mean-square displacement). Higher flexibility results in larger displacements and, eventually, lower electron density. This is because the atoms of a flexible side chain (or other parts of the structure) will be distributed over a larger volume in space, leading to lower density per unit of volume. Based on B-factor values, we can color a protein chain in many graphics programs. Areas with high B-factors are usually red (hot), while low B-factors are blue (cold). Inspecting a PDB structure with such a coloring scheme will immediately reveal highly flexible regions. The molecule's core usually has low B-factors due to the tight packing of the side chains (enzyme active sites are usually located there). The values of the B-factors are generally between 15 to 30 (sq. Angstroms) but often much higher than 30 for flexible regions. More discussion on the B-factor is on the page on structure quality.
The R-factor and resolution are probably the two most central parameters for the assessment of the quality of the structure. The R-factor tells us how good the fit of the structural model to the X-ray data (the electron density) is. A higher resolution of the X-ray data usually provides a better fit and results in a lower R-factor. In my experience, good quality, well-refined protein structures have a resolution of (or better than) 2.2 Å and an R-factor below 20%. At this resolution, the electron density for most atoms looks nice and well-separated from its neighbors.
Below is part of a PDB file header showing some of the data like resolution (2.1 Å), resolution range of the data (from lowest, 29.55 Å to highest, 2.10 Å), the number of reflections collected from the crystal during the X-ray experiment (22179) and the R-factor (0,214). The R-factor is ok but not great in this case. This is due to the high flexibility of parts of the structure, making reliable model building impossible. The result is that parts of the model have not been accounted for, which gives a poor fit to the experimental data.
REMARK 1
REMARK 2
REMARK 2 RESOLUTION. 2.10 ANGSTROMS.
REMARK 3
REMARK 3 REFINEMENT.
REMARK 3 PROGRAM : CNS 1.0
REMARK 3 AUTHORS : BRUNGER,ADAMS,CLORE,DELANO,GROS,GROSSE-
REMARK 3 : KUNSTLEVE,JIANG,KUSZEWSKI,NILGES, PANNU,
REMARK 3 : READ,RICE,SIMONSON,WARREN
REMARK 3
REMARK 3 REFINEMENT TARGET : ENGH & HUBER
REMARK 3
REMARK 3 DATA USED IN REFINEMENT.
REMARK 3 RESOLUTION RANGE HIGH (ANGSTROMS) : 2.10
REMARK 3 RESOLUTION RANGE LOW (ANGSTROMS) : 29.55
REMARK 3 DATA CUTOFF (SIGMA(F)) : 0.000
REMARK 3 DATA CUTOFF HIGH (ABS(F)) : 312841.620
REMARK 3 DATA CUTOFF LOW (ABS(F)) : 0.0000
REMARK 3 COMPLETENESS (WORKING+TEST) (%) : 97.9
REMARK 3 NUMBER OF REFLECTIONS : 22179
REMARK 3
REMARK 3 FIT TO DATA USED IN REFINEMENT.
REMARK 3 CROSS-VALIDATION METHOD : THROUGHOUT
REMARK 3 FREE R VALUE TEST SET SELECTION : RANDOM
REMARK 3 R VALUE (WORKING SET) : 0.214
REMARK 3 FREE R VALUE : 0.247
REMARK 3 FREE R VALUE TEST SET SIZE (%) : 10.000
REMARK 3 FREE R VALUE TEST SET COUNT : 2207
REMARK 3 ESTIMATED ERROR OF FREE R VALUE : 0.005
REMARK 3
Further down, there is a list of the secondary structure elements within the structure, also showing the first and last residue in each element:
HELIX 1 1 PRO A 22 ILE A 26 5 5
HELIX 2 2 GLN A 29 ASP A 42 1 14
HELIX 3 3 PRO A 43 GLY A 46 5 4
HELIX 4 4 ASP A 53 GLY A 57 5 5
HELIX 5 5 SER A 59 LEU A 69 1 11
HELIX 6 6 ASN A 84 ILE A 88 5 5
HELIX 7 7 SER A 114 GLY A 120 1 7
HELIX 8 8 ASP A 123 GLY A 131 1 9
HELIX 9 9 GLY A 138 ASN A 144 1 7
HELIX 10 10 GLU A 152 LEU A 156 5 5
HELIX 11 11 GLU A 157 GLY A 171 1 15
HELIX 12 12 ARG A 202 ASP A 207 1 6
HELIX 13 13 ASP A 220 ASP A 237 1 18
HELIX 14 14 ASP A 237 LEU A 263 1 27
HELIX 15 15 PRO A 264 VAL A 266 5 3
HELIX 16 16 PRO A 269 LEU A 283 1 15
HELIX 17 17 GLY A 287 GLU A 305 1 19
HELIX 18 18 GLY A 311 SER A 324 1 14
HELIX 19 19 HIS A 325 LEU A 327 5 3
HELIX 20 20 VAL A 341 LEU A 349 1 9
SHEET 1 A 5 VAL A 106 LEU A 109 0
SHEET 2 A 5 GLY A 146 ILE A 150 1 O TYR A 147 N VAL A 107
SHEET 3 A 5 PHE A 188 GLY A 194 1 O VAL A 189 N LEU A 148
SHEET 4 A 5 VAL A 48 PHE A 51 1 N VAL A 48 O LEU A 190
SHEET 5 A 5 LEU A 211 GLU A 214 1 O LEU A 211 N LEU A 49
SHEET 1 B 2 ILE A 72 VAL A 75 0
SHEET 2 B 2 VAL A 99 LYS A 102 -1 N ILE A 100 O ALA A 74
SHEET 1 C 2 ALA A 121 LEU A 122 0
SHEET 2 C 2 PHE A 135 GLU A 136 -1 N GLU A 136 O ALA A 121
SHEET 1 D 2 GLU A 172 VAL A 175 0
SHEET 2 D 2 ILE A 182 PRO A 185 -1 O ILE A 182 N VAL A 175
CRYST1 90.259 90.259 83.716 90.00 90.00 120.00 P 65 6
After the general information, the x,y,z coordinates of the atoms are listed:
ATOM 1 N ARG A 18 14.699 61.369 62.050 1.00 39.19 N
ATOM 2 CA ARG A 18 14.500 62.241 60.856 1.00 38.35 C
ATOM 3 C ARG A 18 13.762 61.516 59.729 1.00 36.05 C
ATOM 4 O ARG A 18 14.354 60.740 58.982 1.00 34.91 O
ATOM 5 CB ARG A 18 15.850 62.753 60.334 1.00 42.36 C
ATOM 6 CG ARG A 18 16.537 63.770 61.247 1.00 46.92 C
ATOM 7 CD ARG A 18 17.825 64.314 60.629 1.00 51.24 C
ATOM 8 NE ARG A 18 18.442 65.347 61.462 1.00 54.15 N
When looking at the coordinates, notice that, in this case, the structure starts at amino acid Arg 18! Amino acids from 1 to 17 are missing. The reason, as mentioned above, is poor electron density for these residues, insufficient for building them into the density (see, for example, the discussion on structure quality in homology modeling in a later chapter). Finding the correct positions for the amino acids is impossible without the guiding electron density. This shows that we need to know that many structures in the PDB may have missing parts, sometimes in loop regions. Very often, it is a side chain (or side chains) on the molecule's surface, and in the worst cases, a whole domain may be missing.
The numbers after the first record in the file, ATOM, are just sequential numbers of the atoms in the list. The atom type follows this - for example, CA means C-α, the carbon atom to which the side chain of the amino acid is attached. Next is the main chain carbon atom C, followed by the carbonyl oxygen O. Side chain atoms C-β, CG, and CD (gamma, delta, etc.), are listed according to the Greek alphabet. After the atom type, we find the name of the amino acid (ARG in this case), followed by a letter, in this file A. This letter is the so-called chain identifier. In cases when the structure consists of several polypeptide chains (e.g., as in the tetramers of hemoglobin and Pyruvate kinase discussed earlier), each chain will get its identifier, like A, B, C, etc. The three following numbers (e.g., 14.699, 61.369, 62.050 for the very first atom) are the atom's x,y,z coordinates. As mentioned above, they describe the position of each atom in an orthogonal coordinate system. If we can describe the positions of all atoms in the protein, we can draw the whole molecular structure.
In most cases, the x,y,z coordinates are followed by a number, 1. This is called occupancy. Due to local flexibility, an amino acid's side chain may have two or more different conformations. These conformations can be distinguished in the electron density map of the structure. In this case, the crystallographer will build all conformations and, for each atom, refine a parameter called occupancy (1 for full occupancy, < 1 for partial occupancies, the sum always being 1). In PDB files, these alternate conformations are marked with "ALT."
The B-factor
The numbers in the last column in the file are called the temperature factors or B-factor. The B-factor describes the displacement of the atomic positions from an average (mean) value (mean-square displacement). Higher flexibility results in larger displacements and, eventually, lower electron density. This is because the atoms of a flexible side chain (or other parts of the structure) will be distributed over a larger volume in space, leading to lower density per unit of volume. Based on B-factor values, we can color a protein chain in many graphics programs. Areas with high B-factors are usually red (hot), while low B-factors are blue (cold). Inspecting a PDB structure with such a coloring scheme will immediately reveal highly flexible regions. The molecule's core usually has low B-factors due to the tight packing of the side chains (enzyme active sites are usually located there). The values of the B-factors are generally between 15 to 30 (sq. Angstroms) but often much higher than 30 for flexible regions. More discussion on the B-factor is on the page on structure quality.