Introduction to Protein Secondary Structure: α-Helices and β-Sheets
Here we focus on the general aspects of protein secondary structure. Many of the discussed features are essential for practical applications − for example, in sequence alignment and analysis, homology modeling and model quality analysis, planning mutations, or analyzing protein-ligand interactions.
Helical structures
The most common type of secondary structure in proteins is the α-helix. Linus Pauling was the first to predict the existence of α-helices. The prediction was confirmed when the first three-dimensional structure of a protein, myoglobin (by Max Perutz and John Kendrew) was determined by X-ray crystallography. An example of an α-helix is shown in the image below. This type of protein structure representation is called "stick representation". In the image, only the main chain atoms of the polypeptide are shown connected by sticks, and the side chains are omitted. There are 3.6 residues/turn in an α-helix, which means that there is one residue every 100 degrees of rotation (360/3.6). Each residue is translated 1.5 Å along the helix axis, which gives a vertical distance of 5.4 Å between structurally equivalent atoms in a turn (pitch of a turn). The repeating structural pattern in helices results from similar φ and ψ values. This is reflected in the clustering of the torsion angles within the helical region of the Ramachandran plot. In the section on the Ramachandran plot, we call these regions "energetically most favorable". This means that the particular configuration of the polypeptide chain is such that main chain atoms are packed in an optimal and energetically favorable way avoiding clashes (atoms coming too close to each other).
When looking at the helix in the Figure below, notice how the carbonyl (C=O) oxygen atoms (shown in red) point in one direction towards the amide NH groups four residues away (i, i+4). Together these groups form a hydrogen bond, one of the main forces in stabilizing secondary structures in proteins. The hydrogen bonds are shown as dashed lines.
When looking at the helix in the Figure below, notice how the carbonyl (C=O) oxygen atoms (shown in red) point in one direction towards the amide NH groups four residues away (i, i+4). Together these groups form a hydrogen bond, one of the main forces in stabilizing secondary structures in proteins. The hydrogen bonds are shown as dashed lines.
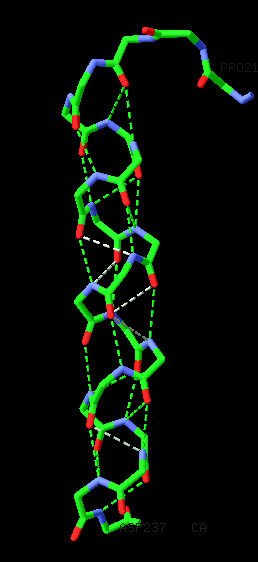
The α-helix is not the only helical structure in proteins. Other helical structures include the 3_10 helix, which is stabilized by hydrogen bonds of the type (i, i+3), the π-helix, which is stabilized by hydrogen bonds of the type (i, i+5), and the left handed L-α helix. The 3_10 helix has a smaller radius than the α-helix, while the π-helix has a larger radius. The first detailed analysis of the occurrence of the π-helix in proteins, based on the analysis of entries in the Protein Data Bank (PDB), was published by Fodje & Al-Karadaghi, 2002. Generally, all these helices are relatively rare when compared to the α-helix.
In addition to the "simple" helical structures mentioned here, there are also so-called coiled-coil structures, in which two or more α-helices build higher-order helical structures together.
In addition to the "simple" helical structures mentioned here, there are also so-called coiled-coil structures, in which two or more α-helices build higher-order helical structures together.
β-strands & β-sheet
The second major secondary structure element in proteins is the β-sheet. β-sheets consist of several β-strands, stretched segments of the polypeptide chain kept together by a network of hydrogen bonds between adjacent strands. An example of a β-sheet, with the stabilizing hydrogen bonds between adjacent strands (dotted lines), is shown in the image below (left):
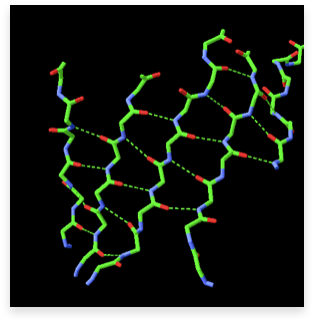
It is important to note that unlike in helices, in which the hydrogen bond donors and acceptors along the helix axis are separated by 4 residues, in β-sheets the hydrogen bond donors and acceptors belong to different strands and may be separated from each other by long segments of the amino acid sequence.
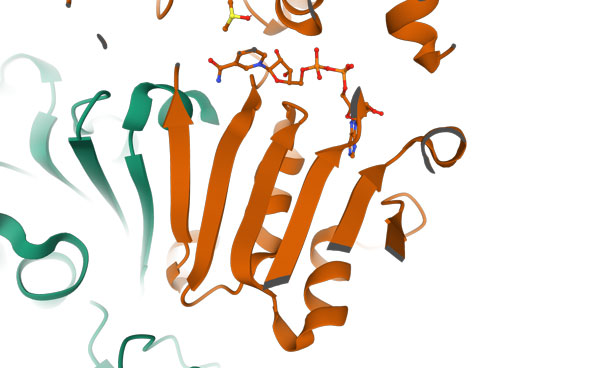
In the upper image on the right, a ribbon presentation of a β-sheet is shown, this time in the context of the 3D structure to which it belongs (the coloring is according to secondary structure). Each β−strand is represented by an arrow, which defines its direction starting from the N- to the C-terminus. When the strand arrows point in the same direction, it is a parallel β-sheet (the PDB code for this structure is 2OHX).
When the strand arrows point in opposite directions, we get an anti-parallel β-sheet (PDB code is 1USR, Newcastle disease virus hemagglutinin-neuraminidase), left image:
In the upper image on the right, a ribbon presentation of a β-sheet is shown, this time in the context of the 3D structure to which it belongs (the coloring is according to secondary structure). Each β−strand is represented by an arrow, which defines its direction starting from the N- to the C-terminus. When the strand arrows point in the same direction, it is a parallel β-sheet (the PDB code for this structure is 2OHX).
When the strand arrows point in opposite directions, we get an anti-parallel β-sheet (PDB code is 1USR, Newcastle disease virus hemagglutinin-neuraminidase), left image:


Loops, turns, and hairpins
When there are only two antiparallel β-strands, like in the Figure on the right, the structural motif is called a β-hairpin. The loop between the two strands is called a β-turn.
Short turns and longer loops are essential in protein 3D structures, connecting strands to strands, strands to α-helices, or helices to helices. The amino acid sequences in loop regions are often highly variable within a protein family. Nevertheless, sometimes, when a loop has some specific function, for example, interaction with another protein, the sequence may be conserved. Loop length in organisms living at elevated temperatures (thermophilic organisms) is usually shorter than in proteins from lower-temperature family members. It presumably gives a protein additional stability at high temperatures, preventing its unfolding and denaturation. During sequence alignment and modeling, when it is essential to have an accurate sequence alignment, the highly variable length of loops justifies the localization of insertions and deletions in the amino acid sequence to loop regions.
The following section will examine how secondary structure elements connect, forming common motifs, folds, and domains.
Short turns and longer loops are essential in protein 3D structures, connecting strands to strands, strands to α-helices, or helices to helices. The amino acid sequences in loop regions are often highly variable within a protein family. Nevertheless, sometimes, when a loop has some specific function, for example, interaction with another protein, the sequence may be conserved. Loop length in organisms living at elevated temperatures (thermophilic organisms) is usually shorter than in proteins from lower-temperature family members. It presumably gives a protein additional stability at high temperatures, preventing its unfolding and denaturation. During sequence alignment and modeling, when it is essential to have an accurate sequence alignment, the highly variable length of loops justifies the localization of insertions and deletions in the amino acid sequence to loop regions.
The following section will examine how secondary structure elements connect, forming common motifs, folds, and domains.